Авторы: Мухин К.Ю., Петрухин А.С.
Часть 1:
- Введение
- Определение, классификация эпилепсии
- Идиопатическая генерализованная эпилепсия
- Детская абсанс эпилепсия.
- Юношеская абсанс эпилепсия.
- Эпилепсия с изолированными генерализованными судорожными приступами.
- Юношеская миоклоническая эпилепсия
- Идиопатическая парциальная эпилепсия
- Роландическая
- Идиопатическая парциальная эпилепсия с затылочными пароксизмами.
Часть 2:
- Криптогенная генерализованная эпилепсия
- Синдром Леннокса-Гасто.
- Эпилепсия с миоклонически-астатическими приступами.
- Эпилепсия с миоклоническими абсансами
- Симптоматическая парциальная эпилепсия
- Симптоматическая височная эпилепсия
- Симптоматическая лобная эпилепсия
- Общие принципы лечения эпилепсии
Синдром Леннокса-Гасто.
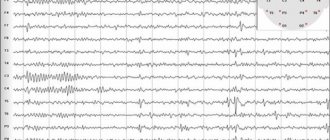
Синдром Леннокса-Гасто (СЛГ) – эпилептическая энцефалопатия детского возраста, характеризующаяся полиморфизмом приступов, специфическими изменениями ЭЭГ и резистентностью к терапии. Частота СЛГ составляет 3-5% среди всех эпилептических синдромов у детей и подростков; болеют чаще мальчики. Заболевание дебютирует, преимущественно, в возрасте 2-8 лет (чаще 4-6 лет). Если СЛГ развивается при трансформации из синдрома Веста, то возможно 2 варианта: Инфантильные спазмы трансформируются в тонические приступы при отсутствии латентного периода и плавно переходят в СЛГ. Инфантильные спазмы исчезают; психомоторное развитие ребенка несколько улучшается; картина ЭЭГ постепенно нормализуется. Затем спустя некоторый латентный промежуток времени, который варьирует у разных больных, появляются приступы внезапных падений, атипичные абсансы и нарастает диффузная медленная пик-волновая активность на ЭЭГ. Для СЛГ характерна триада приступов: пароксизмы падений (атонически- и миоклонически-астатические); тонические приступы и атипичные абсансы. Наиболее типичны приступы внезапных падений, обусловленные тоническими, миоклоническими или атоническими (негативный миоклонус) пароксизмами. Сознание может быть сохранено или выключается кратковременно. После падения не наблюдается судорог, и ребенок сразу же встает. Частые приступы падений приводят к тяжелой травматизации и инвалидизации больных. Тонические приступы бывают аксиальными, проксимальными или тотальными; симметричными либо четко латерализованными. Приступы включают в себя внезапное сгибание шеи и туловища, подъем рук в состоянии полуфлексии или разгибания, разгибание ног, сокращение лицевой мускулатуры, вращательные движения глазных яблок, апноэ, гиперемию лица. Они могут возникать, как в дневное время, так и особенно часто, ночью. Атипичные абсансы также характерны для СЛГ. Проявления их многообразны. Нарушение сознания бывает неполным. Может сохраняться некоторая степень двигательной и речевой активности. Наблюдается гипомимия, слюнотечение; миоклонии век, рта; атонические феномены (голова падает на грудь, рот приоткрыт). Атипичные абсансы обычно сопровождаются понижением мышечного тонуса, что вызывает как бы “обмякание” тела, начиная с мышц лица и шеи. В неврологическом статусе отмечаются проявления пирамидной недостаточности, координаторные нарушения. Характерно снижение интеллекта, не достигающее, однако, тяжелой степени. Интеллектуальный дефицит констатируется с раннего возраста, предшествуя заболеванию (симптоматические формы) или развивается сразу после появления приступов (криптогенные формы). При ЭЭГ-исследовании в большом проценте случаев выявляется нерегулярная диффузная, часто с амплитудной асимметрией, медленная пик-волновая активность с частотой 1,5-2,5 Гц в период бодрствования и быстрые ритмические разряды с частотой около 10 Гц – во время сна. При нейровизуализации могут иметь место различные структурные нарушения в коре головного мозга, включая пороки развития: гипоплазия мозолистого тела, гемимегалэнцефалия, кортикальные дисплазии и пр. В лечении СЛГ следует избегать препаратов, подавляющих когнитивные функции (барбитураты). Наиболее часто при СЛГ применяются вальпроаты, карбамазепин, бензодиазепины и ламиктал. Лечение начинается с производных вальпроевой кислоты, постепенно увеличивая их до максимально переносимой дозы (70-100 мг/кг/сут и выше). Карбамазепин эффективен при тонических приступах – 15-30 мг/кг/сут, но может учащать абсансы и миоклонические пароксизмы. Ряд больных реагирует на увеличение дозы карбамазепина парадоксальным учащением приступов. Бензодиазепины оказывают эффект при всех типах приступов, однако этот эффект временный. В группе бензодиазепинов применяются клоназепам, клобазам (фризиум) и нитразепам (радедорм). При атипичных абсансах может быть эффективен суксилеп (но не как монотерапия). Показана высокая эффективность комбинации вальпроатов с ламикталом (2-5 мг/кг/сут и выше). В США широко используется комбинация вальпроатов с фелбаматом (талокса).
Прогноз при СЛГ тяжелый. Стойкий контроль над приступами достигается лишь у 10-20% больных. Прогностически благоприятно преобладание миоклонических приступов и отсутствие грубых структурных изменений в мозге; негативные факторы – доминирование тонических приступов и грубый интеллектуальный дефицит.
Прогноз
Простые формы миоклонии встречаются у нормальных, здоровых людей и не вызывают серьезных проблем. В некоторых случаях расстройство начинается в одной области тела и распространяется в мышцы в других областях. Более тяжелые случаи миоклонии могут исказить движение и серьезно ограничить способность человека есть, говорить или ходить. Эти типы миоклонии могут указывать на заболевание головного мозга или нервов.
Хотя клоназепам и вальпроат натрия эффективны для лечения большинства случаев расстройства, у некоторых людей на эти препараты возникают побочные реакции. Кроме того, преимущества клоназепама может уменьшаться при длительном приеме.
Эпилепсия с миоклонически-астатическими приступами.
Миоклонически-астатическая эпилепсия (МАЭ) – одна из форм криптогенной генерализованной эпилепсии, характеризующаяся, преимущественно, миоклоническими и миоклонически-астатическими приступами с дебютом в дошкольном возрасте. Дебют МАЭ варьирует от 10 мес. до 5 лет, составляя, в среднем 2,3 года. В 80% случаев начало приступов приходится на возрастной интервал 1-3 года. У подавляющего большинства пациентов заболевание начинается с ГСП с последующим присоединением миоклонических и миоклонически-астатических приступов в возрасте около 4-х лет. Клинические проявления МАЭ полиморфны и включают различные виды приступов: миоклонические, миоклонически-астатические, типичные абсансы, ГСП с возможностью присоединения парциальных пароксизмов. “Ядром” МАЭ являются миоклонические и миоклонически-астатические приступы: короткие, молниеносные подергивания малой амплитуды в ногах и в руках; “кивки” с легкой пропульсией туловища; “удары под колени”. Частота миоклонических приступов высокая, особенно, в утренние часы после пробуждения пациентов. ГСП наблюдаются практически у всех больных, абсансы – у половины. Присоединение парциальных приступов возможно в 20% случаев. При ЭЭГ-исследовании характерно замедление основной активности фоновой записи с появлением генерализованной пик- и полипик-волновой активности с частотой 3Гц. У большинства больных наблюдаются также и региональные изменения: пик-волновая и медленноволновая активность. Лечение начинается с монотерапии препаратами вальпроевой кислоты. Средние дозы – 50-70 мг/кг/сут с постепенным увеличением до 100 мг/кг/сут при отсутствии эффекта. В большинстве случаев эффективна только политерапия: сочетание вальпроатов с ламотриджином или бензодиазепинами или сукцинимидами. Контроль приступов достигается у большинства пациентов, однако, полная ремиссия возможна лишь в 1/3 случаев. Присоединение парциальных пароксизмов значительно ухудшает прогноз; данные приступы наиболее резистентны к терапии. Возможна трансформация МАЭ в синдром Леннокса-Гасто.
Клиническая картина
Первые проявления данного заболевания могут диагностироваться еще до достижения ребенком девяти лет. Однако его дебют приходится на подростковый возраст, что может связываться с происходящими гормональными изменениями.
Первыми проявлениями заболевания являются периодически проявляющиеся краткосрочные асинхронные приступы тонических мышечных сокращений. В большинстве случаев приступы отмечаются утром, непосредственно после пробуждения. Мышечные судороги локализуются в районе верхних конечностей, однако, довольно редко они могут мигрировать на нижние конечности и корпус.
Во время приступа может отмечаться нарушение мелкой моторики, из-за которых пациент теряет возможность осуществлять сложно координированные движения, а в случае локализации приступов в нижних конечностях может наблюдаться нарушение походки или полное отсутствие возможности самостоятельно передвигаться. Миоклоническая эпилепсия может иметь как единичный, так и кластерный характер.
Менее чем в 5 % случаев юношеская эпилепсия может протекать с проявлением только миоклонических приступов. Однако практически всегда после проявления миоклонических парадигм течение заболевания осложняется появлением генерализованных тонико-клинических эпилептических приступов. Кроме этого примерно у половины больных со временем проявляются абсансы – кратковременные потери сознания.
Эпилепсия с миоклоническими абсансами.
Эпилепсия с миоклоническими абсансами (ЭМА) – форма абсансной эпилепсии, характеризующаяся частыми приступами абсансов, протекающих с массивными миоклониями мышц плечевого пояса и рук, и резистентностью к терапии. Дебют приступов при ЭМА варьирует от 1 до 7 лет (в среднем, 4 года); преобладают по полу мальчики. Миоклонические абсансы у большинства больных являются первым видом приступов. В некоторых случаях заболевание может начинаться с ГСП с последующим присоединением абсансов. Сложные абсансы с массивным миоклоническим компонентом составляют “ядро” клинической картины ЭМА. Типичны абсансы с интенсивными миоклоническими подергиваниями плечевого пояса, плеч и рук, носящими, обычно, билатерально-синхронный и симметричный характер. При этом может наблюдаться легкий наклон туловища и головы кпереди (пропульсия), отведение и приподнимание плеч (тонический компонент). У большинства пациентов также отмечаются миоклонические подергивания мышц шеи (короткие серийные кивки), синхронно с подергиваниями плеч и рук. Характерна высокая частота абсансов, достигающая 10 приступов в час и более. Продолжительность приступов составляет от 5 до 30 сек, причем, характерны длительные абсансы – более 10 сек. Приступы нередко учащаются в утренние часы. Основным фактором, провоцирующим возникновение абсансов при ЭМА, является гипервентиляция. В большинстве случаев (80%) абсансы сочетаются с генерализованными судорожными приступами. Характерна редкая частота ГСП, обычно, не превышающая 1 раза в мес. Изменения ЭЭГ в межприступном периоде выявляются практически во всех случаях. Замедление основной активности фоновой записи отмечается нечасто, главным образом, у пациентов с интеллектуальным дефицитом. Типичный ЭЭГ-паттерн – генерализованная пик- (или, реже, полипик- ) волновая активность с частотой 3 Гц. Достоверным для установления диагноза ЭМА является появление разрядов при электромиографии в ответ на миоклонические сокращения мышц, возникающих синхронно с пик-волновой активностью на ЭЭГ (полиграфическая запись). Лечение. Стартовое лечение осуществляется с монотерапии препаратами, производными вальпроевой кислоты. Средняя дозировка 50-70 мг/кг/сут; при хорошей переносимости – постепенное увеличение до 80-100 мг/кг/сут. В большинстве случаев монотерапия уряжает приступы, но не приводит к достаточному контролю над ними. В этом случае рекомендуется сочетать прием вальпроатов с сукцинимидами или ламотриджином.
Практически у всех больных при применении политерапии в адекватно высоких дозировках удается добиться хорошего контроля над приступами, однако, ремиссия возникает лишь в 1/3 случаев. У большинства пациентов имеются серьезные проблемы с социальной адаптацией.
Что такое миоклония?
Миоклония (или миоклонус) — это симптом, а не заболевание, характеризующаяся кратковременным быстрым сокращениям мышц или группы мышц.
Миоклонические подёргивания или толчки обычно вызываются внезапными сокращениями мышц, называемыми положительным миоклонусом, или мышечным расслаблением, называемым отрицательным миоклонусом. Миоклонические подёргивания могут происходить поодиночке или последовательно, по шаблону или без шаблона. Они могут происходить нечасто или многократно каждую минуту. Миоклония иногда возникает в ответ на внешнее событие или когда человек пытается сделать движение. Человек, испытывающий подёргивания, не может контролировать их.
В своей простейшей форме миоклония состоит из подергивания мышц с последующим расслаблением. В пример можно привести икоту, что тоже является миоклонией. Другими знакомыми примерами состояния являются подергивания, которые некоторые люди испытывают во время засыпания. Эти простые формы миоклонии встречаются у нормальных, здоровых людей и не вызывают затруднений. При более широком распространении состояние может вызывать постоянные шокоподобные сокращения в группе мышц.
В некоторых случаях миоклония начинается в одной области тела и распространяется в мышцы в других областях. Более тяжелые случаи состояния могут исказить движение и серьезно ограничить способность человека есть, говорить или ходить. Эти типы миоклонии могут указывать на основное заболевание мозга или нервов.
Диагноз устанавливают на основании признаков и иногда подтверждают результатами электромиографического исследования. Лечение включает в себя коррекцию обратимых заболеваний головного мозга или нервной системы и симптоматическую терапию лекарственными препаратами.
Симптоматическая парциальная эпилепсия
При симптоматических парциальных формах эпилепсии выявляются структурные изменения в коре головного мозга. Причины, детерминирующие их развитие, многообразны и могут быть представлены двумя основными группами: перинатальными и постнатальными факторами. Верифицированное перинатальное поражение ЦНС в анамнезе констатируется у 35% пациентов (внутриутробные инфекции, гипоксия, эктомезодермальные дисплазии, кортикальные дисплазии, родовая травма и пр.). Среди постнатальных факторов следует отметить нейроинфекции, черепно-мозговые травмы, опухоли коры головного мозга. Дебют приступов при симптоматической парциальной эпилепсии варьирует в широком возрастном диапазоне, с максимумом в предшкольном возрасте. Для этих случаев характерно наличие изменений в неврологическом статусе, нередко в сочетании со снижением интеллекта; появление региональных паттернов на ЭЭГ, резистентность приступов к АЭП и возможность хирургического лечения. Выделяют симптоматические парциальные формы эпилепсии: височную, лобную, теменную и затылочную. Две первых – наиболее частые и составляют до 80% всех случаев.
Симптомы миоклонии
Миоклония может быть слабой или тяжелой. Мышцы могут сокращаться быстро или медленно, ритмично и неритмично. Миоклонические подергивания могут быть редкими или частыми. Они могут наступать спонтанно или под действием внезапного шума, света или движения. Например, их можно спровоцировать, потянувшись за каким-нибудь предметом или делая шаг. При болезни Крейтцфельдта–Якоба (редкое дегенеративное заболевание головного мозга) миоклония усиливается при внезапном испуге.
Миоклония, развившаяся в результате нарушения метаболизма, может быть длительной и затрагивать разные группы мышц, иногда приводя к судорогам.
Симптоматическая височная эпилепсия.
Клинические проявления симптоматической височной эпилепсии (ВЭ) крайне разнообразны. В ряде случаев атипичные фебрильные судороги предшествуют развитию заболевания. ВЭ проявляется простыми, сложными парциальными, вторично-генерализованными приступами или их сочетанием. Особенно характерно наличие сложных парциальных приступов, протекающих с расстройством сознания, в сочетании с сохранной, но автоматизированной двигательной активностью. Автоматизмы при сложных парциальных приступах могут быть односторонними, возникая на гомолатеральной очагу стороне, и часто сочетаются с дистонической установкой кисти на контрлатеральной стороне. ВЭ подразделяют на амигдало-гиппокампальную (палеокортикальную) и латеральную (неокортикальную) эпилепсию. Амигдало-гиппокампальная ВЭ характеризуется возникновением приступов с изолированным расстройством сознания. Наблюдается застывание больных с маскообразным лицом, широко раскрытыми глазами и уставленным в одну точку взглядом (он как бы «таращится» – «staring» в англоязычной литературе). При этом могут констатироваться различные вегетативные феномены: побледнение лица, расширение зрачков, потливость, тахикардия. Выделяют 3 типа СПП с изолированным расстройством сознания: 1.Выключение сознания с застыванием и внезапным прерыванием двигательной и психической активности. 2.Выключение сознания без прерывания двигательной активности. 3.Выключение сознания с медленным падением (“обмяканием”) без судорог (“височные синкопы”). Характерны также вегетативно-висцеральные пароксизмы. Приступы проявляются ощущением абдоминального дискомфорта, болей в области пупка или эпигастриума, урчания в животе, позывов на дефекацию, отхождения газов (эпигастральные приступы). Возможно появление “восходящего эпилептического ощущения”, описываемого больными как боль, изжога, тошнота, исходящего из живота, и поднимающегося к горлу, с чувством сжатия, сдавления шеи, комка в горле, нередко с последующим выключением сознания и судорогами. При вовлечении в процесс миндалевидного комплекса возникают приступы страха, паники или ярости; раздражение крючка вызывает появление обонятельных галлюцинаций. Возможны приступы с нарушением психических функций (сновидные состояния, уже виденного или никогда не виденного и пр.). Латеральная ВЭ проявляется приступами с нарушением слуха, зрения и речи. Характерно появление ярких цветных структурных (в отличии от затылочной эпилепсии) зрительных галлюцинаций, а также сложных слуховых галлюцинаций. Около 1/3 женщин, страдающих ВЭ, отмечают учащение приступов в перименструальном периоде. При неврологическом обследовании детей, страдающих ВЭ, нередко обнаруживаются микроочаговые симптомы, контрлатерально очагу: недостаточность функции 7 и 12 пары черепных нервов по центральному типу, оживление сухожильных рефлексов, появление патологических рефлексов, легкие координаторные расстройства и пр. С возрастом у большинства пациентов выявляются стойкие нарушения психики, проявляющиеся, преимущественно, интеллектуально-мнестическими или эмоционально-личностными расстройствами; характерно появление выраженных расстройств памяти. Сохранность интеллекта зависит, главным образом, от характера структурных изменений в мозге. При ЭЭГ-исследовании наблюдается пик-волновая или чаще стойкая региональная медленноволновая (тета) активность в височных отведениях, обычно с распространением кпереди. У 70% больных выявляется выраженное замедление основной активности фоновой записи. У большинства пациентов, с течением времени, эпилептическая активность отмечается битемпорально. Для выявления очага, локализованного в медио-базальных отделах, предпочтительнее применение инвазивных сфеноидальных электродов. При нейрорадиологическом обследовании обнаруживаются различные макроструктурные нарушения в головном мозге. Частой находкой при МРТ исследовании является медиальный височный (инцизуральный) склероз. Нередко также отмечается локальное расширение борозд, уменьшение в объеме вовлеченной височной доли, парциальная вентрикуломегалия. Лечение ВЭ – сложная задача; многие пациенты резистентны к терапии. Базовые препараты – производные карбамазепина. Средняя суточная дозировка составляет 20 мг/кг. При неэффективности – увеличение дозы до 30-35 мг/кг/сут и выше до появления положительного эффекта или первых признаков интоксикации. При отсутствии эффекта следует отказаться от применения карбамазепина, назначив вместо него дифенин при сложных парциальных приступах или вальпроаты при вторично – генерализованных пароксизмах. Дозировка дифенина в сутки при лечении ВЭ составляет 8-15 мг/кг, вальпроатов – 50-100 мг/кг/сут. При отсутствии эффекта от монотерапии возможно применение политерапии: финлепсин + депакин, финлепсин + фенобарбитал, финлепсин + ламиктал, фенобарбитал + дифенин (последняя комбинация вызывает значительное снижение внимания и памяти, особенно у детей). В дополнение к базовой антиконвульсантной терапии у женщин могут применяться половые гормоны, особенно эффективные при менструальной эпилепсии. Применяется оксипрогестерона капронат 12,5% раствор 1-2 мл в/м однократно на 20-22 день менструального цикла. Прогноз зависит от характера структурного поражения мозга. С возрастом у большинства пациентов нарастают стойкие нарушения психики, значительно затрудняющие социальную адаптацию. В целом, около 30% больных, страдающих ВЭ, являются резистентными к традиционной противосудорожной терапии и являются кандидатами на нейрохирургическое вмешательство.
Лечение миоклонии
Лечение миоклонии направлено на лекарства, которые могут помочь уменьшить симптомы. Препаратом первого выбора для лечения, особенно определенных видов миоклонии действия, является клоназепам, тип транквилизатора. Дозировки клоназепама обычно увеличивают постепенно, пока больной не почувствует улучшений или не появятся побочные эффекты. Сонливость и потеря координации являются общими побочными эффектами. Благоприятные эффекты клоназепама могут со временем уменьшаться, если у человека разовьется толерантность к препарату.
Многие лекарства, используемые для лечения миоклонии, такие как барбитураты, леветирацетам, фенитоин и примидон, также используются для лечения эпилепсии. Барбитураты замедляют центральную нервную систему и вызывают транквилизирующий или антисейзерный эффект. Фенитоин, леветирацетам и примидон являются эффективными противоэпилептическими препаратами, хотя фенитоин может вызывать печеночную недостаточность или оказывать другие вредные долгосрочные эффекты. Вальпроат натрия является альтернативной терапией миоклонуса и может использоваться как один, так и в сочетании с клоназепамом. Хотя клоназепам и/или вальпроат натрия эффективны для большинства людей с расстройством, у некоторых людей возникают побочные реакции на эти препараты.
Симптоматическая лобная эпилепсия.
Клиническая симптоматика лобной эпилепсии (ЛЭ) разнообразна. Заболевание проявляется простыми и сложными парциальными приступами, а также, что особенно характерно, вторично генерализованными пароксизмами. Выделяют следующие формы ЛЭ: моторная, оперкулярная, дорсолатеральная, орбитофронтальная, передняя фронтополярная, цингулярная, исходящая из дополнительной моторной зоны. Моторные пароксизмы возникают при раздражении передней центральной извилины. Характерны джексоновские приступы, развивающиеся контрлатерально очагу. Судороги имеют преимущественно клонический характер и могут распространяться по типу восходящего (нога — рука — лицо) или нисходящего (лицо — рука — нога) марша; в некоторых случаях со вторичной генерализацией. При очаге в парацентральных дольках судороги могут наблюдаться в ипсилатеральной конечности или билатерально. Постприступная слабость в конечностях (паралич Тодда) – нередкий феномен ЛЭ. Оперкулярные приступы возникают при раздражении оперкулярной зоны нижней лобной извилины на стыке с височной долей. Проявляются пароксизмами жевательных, сосательных, глотательных движений, причмокиванием, облизыванием, покашливанием; характерна гиперсаливация. Возможно ипсилатеральное подергивание мышц лица, нарушение речи или непроизвольная вокализация. Дорсолатеральные приступы возникают при раздражении верхней и нижней лобной извилин. Проявляются адверсивными приступами с насильственным поворотом головы и глаз, обычно контрлатерально очагу раздражения. При вовлечении задних отделов нижней лобной извилины (центр Брока) констатируются пароксизмы моторной афазии. 0рбитофронтальные приступы возникают при раздражении орбитальной коры нижней лобной извилины и проявляются разнообразными вегетативно-висцеральными феноменами. Характерны эпигастральные, кардиоваскулярные (боли в области сердца, изменение сердечного ритма, артериального давления), респираторные (инспираторная одышка, чувство удушья, сжатия в области шеи, «кома» в горле) приступы. Нередко появляются фаринго-оральные автоматизмы с гиперсаливацией. Обращает на себя внимание обилие вегетативных феноменов в структуре приступов: гипергидроз, бледность кожи, нередко с гиперемией лица, нарушение терморегуляции и пр. Возможно появление типичных сложных парциальных (психомоторных) пароксизмов с автоматизмами жестов.
Передние (фронтополярные) приступы возникают при раздражении полюса лобных долей. Характеризуются простыми парциальными приступами с нарушением психических функций. Проявляются ощущением внезапного «провала мыслей», «пустоты в голове», растерянности или, наоборот, насильственным воспоминанием; мучительным, тягостным ощущением необходимости что-то вспомнить. Возможен насильственный «наплыв мыслей», «вихрь идей» – ощущение внезапного появления в сознании мыслей, не связанных по содержанию с текущей мыслительной деятельностью. Пациент не имеет возможности избавиться от этих мыслей до окончания приступа. Цингулярные приступы исходят из передней части поясной извилины медиальных отделов лобных долей. Проявляются, преимущественно, сложными, реже простыми парциальными приступами с нарушением поведения и эмоциональной сферы. Характерны сложные парциальные приступы с автоматизмами жестов, покраснением лица, выражением испуга, ипсилатеральными моргательными движениями, а иногда, клоническими судорогами контрлатеральных конечностей. Возможно появление пароксизмальных дисфорических эпизодов со злобностью, агрессивностью, психомоторным возбуждением. Приступы, исходящие из дополнительной моторной зоны впервые были описаны Реnfield, но систематизированы лишь в последнее время. Это довольно частый вид приступов, особенно, если учесть, что пароксизмы, возникающие в других отделах лобной доли, нередко иррадиируют в дополнительную моторную зону. Характерно наличие частых, обычно ночных, простых парциальных приступов с альтернирующими гемиконвульсиями, архаическими движениями; приступов с прекращением речи, нечеткими плохо локализованными чувствительными ощущениями в туловище и конечностях. Парциальные моторные приступы проявляются обычно тоническими судорогами, возникающими то с одной, то с другой стороны, или билатерально (при этом выглядят как генерализованные). Характерно тоническое напряжение с подъемом контрлатеральной руки, адверсией головы и глаз (больной как бы смотрит на свою поднятую руку). Описано возникновение “тормозных” приступов с пароксизмальным гемипарезом. Приступы архаических движений возникают обычно в ночное время с высокой частотой (до 3-10 раз за ночь, нередко, каждую ночь). Характеризуются внезапным пробуждением пациентов, криком, гримасой ужаса, двигательной бурей: размахиванием руками и ногами, боксированием, педалированием (напоминающим езду на велосипеде), тазовыми движениями (как при коитусе) и пр. Степень нарушения сознания флюктуирует, но в большинстве случаев сознание сохранено. Данные приступы следует дифференцировать от истерических и пароксизмальных ночных страхов у детей. ЭЭГ-исследование при ЛЭ может показывать следующие результаты: норма, пик-волновая активность или замедление (периодическое ритмическое или продолженное) регионально в лобных, лобно-центральных или лобно-височных отведениях; бифронтальные независимые пик-волновые очаги; вторичная билатеральная синхронизация; региональная фронтальная низкоамплитудная быстрая (бетта) активность. 0чаги, локализованные в орбитофронтальной, оперкулярной и дополнительной моторной зоне могут не давать изменений при наложении поверхностных электродов и требуют применения глубинных электродов или кортикографии. При поражении дополнительной моторной зоны, ЭЭГ-паттерны нередко ипсилатеральны приступам или билатеральны, или имеет место феномен вторичной билатеральной синхронизации Джаспера. Лечение ЛЭ осуществляется по общим принципам терапии локализационно-обусловленных форм эпилепсии. Карбамазепин (20-30 мг/кг/сут) и вальпроаты (50-100 мг/кг/сут) являются препаратами выбора; дифенин, барбитураты и ламотриджин – резервными. Вальпроаты особенно эффективны в случае вторично-генерализованных судорожных приступов. При неэффективности монотерапии применяется политерапия – комбинация перечисленных выше препаратов. Полная резистентность приступов к проводимой терапии АЭП является поводом для рассмотрения вопроса об оперативном вмешательстве. Прогноз ЛЭ зависит от характера структурного поражения мозга. Частые приступы, резистентные к терапии, значительно ухудшают социальную адаптацию больных. Приступы, исходящие из дополнительной моторной зоны, обычно резистентны к традиционным АЭП и требуют хирургического лечения.
Диагноз
Диагноз ставится на основании сочетания в клин, картине миоклонического гиперкинеза с эпилептическими припадками. Для диагностики важное значение имеет семейный характер заболевания. Дифференциальный диагноз проводится с весьма сходной мозжечковой миоклонической диссинергией Ханта — наследственным, аутосомно-рецессивным заболеванием, при к-ром также наблюдаются миоклонический гиперкинез и эпилептические припадки, но, в отличие от М.-э., мозжечковые расстройства гораздо более выражены. В отличие от М.-э., при кожевниковской эпилепсии (см.) клонические судороги наблюдаются в определенной части тела и обычно не носят генерализованного характера. Особенно трудно установить ранний диагноз в спорадических случаях, когда заболевание проявляется неполным клин, синдромом — или эпилептическими припадками, или миоклоническим гиперкинезом. В этом случае имеет значение семейный анамнез, а также уменьшение содержания мукополисахаридов в сыворотке крови.
Общие принципы лечения эпилепсии
В настоящее время выработаны общепринятые международные стандарты по лечению эпилепсии, которые необходимо соблюдать для повышения эффективности лечения и улучшения качества жизни пациентов.
Лечение эпилепсии может быть начато только после установления точного диагноза. Термины “предэпилепсия” и “профилактическое лечение эпилепсии” являются абсурдными. Существуют две категории пароксизмальных неврологических расстройств: эпилептические и неэпилептические (обмороки, снохождения, ночные страхи и пр.), и назначение АЭП оправдано только в случае болезни. По мнению большинства неврологов, лечение эпилепсии следует начинать после повторного приступа. Единичный пароксизм может быть “случайным”, обусловленным лихорадкой, перегревом, интоксикацией, метаболическими расстройствами и не относиться к эпилепсии. В этом случае немедленное назначение АЭП не может быть оправданным, так как данные препараты являются потенциально высокотоксичными и не применяются с целью “профилактики”. Таким образом, АЭП могут применяться только в случае повторных непровоцируемых эпилептических приступов (т.е. при эпилепсии по определению).
В случае установления точного диагноза эпилепсии необходимо решить вопрос, следует или нет назначать АЭП? Разумеется, в подавляющем большинстве случаев, АЭП назначаются немедленно после диагностирования эпилепсии. Однако при некоторых доброкачественных эпилептических синдромах детского возраста (прежде всего, при роландической эпилепсии) и рефлекторных формах болезни (эпилепсия чтения, первичная фотосенситивная и др.), допускается ведение пациентов без применения АЭП. Подобные случаи должны быть строго аргументированы.
Диагноз эпилепсии установлен и решено назначить АЭП. С 1980-х годов в клинической эпилептологии прочно утвердился принцип монотерапии: купирование эпилептических приступов должно осуществляться преимущественно одним препаратом. С появлением хроматографических методов определения уровня АЭП в крови стало очевидным, что многие антиконвульсанты имеют взаимный антагонизм, и одновременное их применение может значительно ослабить противосудорожный эффект каждого. Кроме того, применение монотерапии позволяет избежать возникновения тяжелых побочных эффектов и тератогенного воздействия, частота которых значительно возрастает при назначении нескольких препаратов одновременно. Таким образом, в настоящее время полностью доказана несостоятельность старой концепции о назначении большого количества АЭП одновременно в малых дозах. Политерапия оправдана только в случае резистентных форм эпилепсии и не более 3-х АЭП одновременно.
Подбор АЭП не должен быть эмпирическим. АЭП назначаются строго в соответствии с формой эпилепсии и характером приступов. Успех лечения эпилепсии во многом определяется точностью синдромологической диагностики (табл. 3).
АЭП назначаются, начиная с малой дозы, с постепенным увеличением до достижения терапевтической эффективности или появления первых признаков побочных эффектов. При этом определяющим является клиническая эффективность и переносимость препарата, а не содержание его в крови (табл. 4).
В случае неэффективности одного препарата, он должен быть постепенно заменен другим АЭП, эффективным при данной форме эпилепсии. При неэффективности одного АЭП нельзя сразу прибавлять к нему второй препарат, то есть переходить на политерапию не используя всех резервов монотерапии.
Принципы отмены АЭП.
АЭП могут быть отменены спустя 2,5-4 года полного отсутствия приступов. Клинический критерий (отсутствие приступов) является основным критерием отмены терапии. При большинстве идиопатических форм эпилепсии отмена препаратов может осуществляться через 2,5 (роландическая эпилепсия) — 3 года ремиссии. При тяжелых резистентных формах (синдром Леннокса-Гасто, симптоматическая парциальная эпилепсия), а также при юношеской миоклонической эпилепсии, данный период увеличивается до 3-4 лет. При продолжительности полной терапевтической ремиссии в течение 4-х лет, лечение должно быть отменено во всех случаях. Наличие патологических изменений на ЭЭГ или пубертатный период пациентов не являются факторами, задерживающими отмену АЭП при отсутствии приступов более 4-х лет. Не существует единого мнения по вопросу о тактике отмены АЭП. Лечение может быть отменено постепенно в течение 1-6 мес или одномоментно по усмотрению врача.
часть-1 часть-2
Лечение и Прогноз
Лечение симптоматическое. Применяется противосудорожная терапия, назначают фенобарбитал, бензонал, седуксен, хлоралгидрат. Последние два средства могут значительно уменьшить на короткое время миоклонический гиперкинез. Применение хлоралгидрата может вызвать привыкание к нему и быстрое развитие кахексии, Показаны также повторные длительные курсы лечения глутаминовой к-той.
Прогноз неблагоприятный. Средняя продолжительность болезни ок. 20 лет, нек-рые больные доживают до старческого возраста. Наиболее часто причиной смерти является нарастающая кахексия, нередко пневмонии и другие интеркуррентные заболевания.
Супругам, в семье к-рых есть больной Миоклонус-эпилепсией, для решения вопроса о рождении ребенка рекомендуется обратиться в медико-генетическую консультацию (см.).
Библиография:
Давиденков С. Н. Миоклонус-эпилепсия, в кн.: Неврол. и генетика, под ред. С. Н. Давиденкова, т. 2, с. 195, М., 1936; Дзержинский В. Myoclonia Unverricht’a, Журн, невропат, и психиат., кн. 5-6, с. 293, 1910; Маккьюсик В. А. Наследственные признаки человека, пер. с англ., М., 1976; Мельников С. А. Миоклонус-эпилепсия как синдром при некоторых заболеваниях головного мозга, Журн, невропат, и психиат., т. 57, в. 6, с. 740, 1957, библиогр.; de Ajuriaguerra J., Sigwald J. et Piot C. Myoclonie-epilepsie familiale de type Unverricht, Presse med., p. 1813, 1954; Bogaert L. Sur l’epilepsie-myoclonie progressive d’Unverricht — Lundborg, Mschr. Psychiat. Neurol., Bd 118, S. 170, 1949, Bibliogr.; Davison Ch. a. Keshner M. Myoclonus epilepsy, Arch. Neurol. Psychiat. (Chic.), v. 43, p. 524, 1940; Gambetti P. a. o. Myoclonic epilepsy with lafora bodies, Arch. Neurol. (Chic.), v. 25, p. 483, 1971; Hallidaу A. M. Les differents types des myoclonies, Rev. neurol., t. 119, p. 135, 1968; Handbook of clinical neurology, ed. by P. J. Yinken a. G. W. Bruyn, v. 15, p. 121, 1974, v. 27, p. 171, Amsterdam a. o., 1976; Lafora G. R. u. Glueck B. Beitrag zur Histopathologie der myoclonischen Epilepsie, Z. ges. Neurol. Psychiat., Bd 6, S. 1, 1911, Bibliogr.; LundborgH. Die progressive Myoclonus-Epilepsie, Up-sala, 1903; Unverricht H. Die Myoclonie, L]3z.— Wien, 1891.
P. А. Ткачев.