Prion diseases - transmissible spongiform encephalopathies (TSEs) - are a relatively new group of degenerative diseases of the central nervous system. The disorders are characterized by a chronic, unusual interaction between the immune system and the presumed causative factor, the prion protein. In the peripheral path of the disease, it multiplies in the endoretular system, from where it passes into the central nervous system (brain and spinal cord), where it causes vascularization of neurocytes, glial cells, spongiomatous reconstruction of the neuropilus, extinction and reduction in the number of neuronal cells, gliosis without significant involvement of inflammation.
“Bio/mol/text”-2012
This article was submitted to the competition of popular science works “bio/mol/text”-2012 in the category “Best News Message”.
The competition is sponsored by the visionary Thermo Fisher Scientific.
It all started when in the 20th century scientists became interested in the nature of unusual diseases in humans and animals: kuru, Creutzfeldt-Jakob, scrapie. The noticeable similarity in the pathology of these diseases gave rise to the hypothesis of their infectivity, which was subsequently confirmed experimentally. Then the question arose about the causative agent of these diseases. Before the answer was found, the extraordinary properties of the pathogens were revealed: they do not reproduce on artificial nutrient media, are resistant to high temperature, formaldehyde, various types of radiation, and the action of nucleases. Purification of the infectious material and its study made it possible to proclaim that “everything is to blame” for the protein, which 30 years ago received the name prion (from the English prion - protein infection).
Thus, famous American scientists - virologist and doctor D.K. Gaidushek, who discovered the infectious nature of prion diseases, in 1976, and biochemist S.B. Prusiner, who identified prions and developed the prion theory in 1997, were awarded Nobel Prizes. Their work became the impetus for subsequent research, thanks to which new types of prion infections were studied. But, even despite the undying interest in the “prion topic,” the formation of prions remains a mystery to this day.
How to slow down the progression of prion disease
Until recently, it was difficult to slow down prion disease, let alone cure it.
Modern neurosurgery can alleviate degenerative complications by transplanting neurons for prion disease.
A revolutionary attempt to stop pathogenic proteins was pioneered by a team of doctors from the University of Pittsburgh. Dr. Kondziolka's team transplanted neurons in vitro (2-6 million LBS neurons) into 12 patients as part of the study. 12 months after surgery, none of the recipients had any adverse effects from the transplant, and all received new cells. In 6 patients there was a significant improvement in condition.
Several years ago, the journal Nature reported on the development of antibodies against prions in England by a team of scientists led by Simone Hawke (Imperial College). In tests on infected mice, rodents that received the antibodies were cured within a month of infection.
Doctors intend to use this opportunity to clear neurons of pathogenic proteins to treat a new variant of Creutzfeldt-Jacobs disease in humans. Further research should help improve drugs that can clear prions from the brain and its microglia.
Recently, M. Pfeiffer and several other German scientists discovered and confirmed a method to clear the brain of prions. They proved that RNA interference can successfully treat diseases by removing from the RNA cell the part responsible for the production of pathogenic proteins. This prevented them from mutating into dangerous proteins.
A similar method is the effect of artificially synthesized primers (oligonucleotides) against prions. Like RNA interference, primer design is under investigation.
Theoretically, the PrP13 peptide, similar to the structural product found in these diseases, may be promising in the treatment of diseases. The substance targets the PRNP gene, which should affect the conversion of PrPC to PrPres. The last pathogenic molecule named changes to a PrPC-like structure. The substance goes by the acronym PrP13, and animal studies have been able to prolong survival by 50-300%.
Biological essence of prions
Figure 1. The metaphor for neurodegenerative brain damage is that neural tissue becomes a sponge as a result of massive neuronal death.
The prion molecule is not something exotic: in a “normal” form it is present on the surface of the nerves of each of us. At the same time, we feel great, and our nerve cells are alive and healthy. However, this is all until our normal protein “degenerates” into an abnormal form. And if this happens, it will lead to terrifying consequences: the infectious form of prions has the ability to “stick together” with other molecules and, moreover, “convert” them into the same form, causing a “molecular epidemic”. As a result of this polymerization, toxic protein plaques appear on the nerve cell, and it dies [1]. In place of the dead cell, a void is formed - a vacuole filled with liquid. Over time, one neuron after another will disappear, and more and more “holes” will form in the brain, until finally the brain turns into a sponge (Fig. 1), which will inevitably be followed by death.
There is a simplified idea that polymerized prion fibrils “puncture” the neuron, causing its death. In fact, this is not entirely true: spherical prion aggregates preceding the fibrillar stage are also toxic (at least for Alzheimer's disease): “Alzheimer's neurotoxin: not only fibrils are poisonous.” - Ed.
But how can a normal natural protein (denoted PrPC) suddenly become pathological (PrPSc; Sc - from the word “scrapie”)? What is going to happen? As in the case of a “normal” infection, such transformation requires an encounter with an infectious prion molecule. There are two ways of transmitting this molecule: hereditary (due to mutations in the gene encoding the protein) and infectious. That is, the introduction of a prion can occur unexpectedly - for example, when eating insufficiently well-fried or cooked meat (which should contain the PrPSc form), during a blood transfusion, during organ and tissue transplantation, or when administering pituitary hormones of animal origin.
And then an amazing event occurs: normal protein molecules, in contact with pathological ones, themselves transform into them, changing their spatial structure (the mechanism of transformation remains a mystery to this day) [1]. Thus, the prion, as a real infectious agent, infects normal molecules, triggering a chain reaction that is destructive to the cell.
Individual nosological units
Human prion diseases are species specific. Basically, they spread only among individuals of one species. But scientists have demonstrated interspecies transmission (overcoming the interspecies barrier): this occurred in scrapie sheep with its transmission to cows (mad cow disease). Transmission of the disease from animals to humans, despite many suspicions, has not been demonstrated.
A number of diseases are of hereditary origin, some are infectious. Sometimes the disease occurs unexpectedly, without the possibility of prevention.
Kuru
The disease existed only in Papua New Guinea in tribes of cannibals who consumed the brains of the dead. It manifests itself as tremor (“kuru” means “tremor”), speech disturbances, and balance disorders (ataxia). Death occurs a few months later. After stopping cannibalism, the kuru disease disappeared.
Creutzfeldt-Jakob disease
It is a rare disorder that affects older people (most often 50-70 year olds). Its course is relatively rapid, death occurs within a few months (up to a year) from the onset of the first symptoms. Neurons quickly die, and progressive dementia develops, which has no known cure. There is some hereditary predisposition to this disease:
- Sporadic form . The incidence is 1-2/1000000. Problems begin around age 65. The disorder occurs in the form of acute progression of dementia (over 2-3 months), ataxia, and myoclonus. There are no ways to prolong the life of neurons with this prion disease; the patient dies 5-12 months after the appearance of the first symptoms.
- Iatrogenic form . It occurred in patients receiving human growth hormone from cadaveric pituitary glands (today it is prepared recombinantly), transplantation of hard membranes, pericardium, and cornea. There is a risk of neurosurgical transmission. Infection is also possible through transfusion.
- Family form . This is a genetic form with a mutation in the PRNP gene and neuropsychiatric symptoms.
- New option . Characterized by psychiatric symptoms (anxiety, depression, behavioral changes), progressive cerebellar syndrome, myoclonus, and other neurological symptoms. Unlike the sporadic form, the new variant is characterized by a slow progression of viral infection; prion disease affects younger age groups. Transmission occurs through nutritional means (through consumption of meat from affected animals). The incubation period is more than 10 years. Around 200 people have died worldwide to date.
- The childhood version of Creutzfeldt-Jakob disease is Alper disease . Has the same manifestations as the disease in adults; in addition accompanied by hepatic steatosis.
Gerstmann-Sträussler-Scheinker disease
Gerstmann-Strüssler-Scheinker disease is a very rare autosomal dominant hereditary disease that begins in the 3rd-4th decade of life. It manifests itself as a slow slowdown in the functioning of neurons by prions, and progressive dysfunction of the cerebellum and spinal cord. Signs:
- ataxia;
- pyramidal symptoms;
- dysarthria;
- dysphagia;
- gradually developing dementia.
At the beginning of the disorder, apathy and depression may occur. The average duration of the disease is about 4-5 years. Several PRNP mutations associated with this disorder have been described.
Fatal familiar insomnia
This is a very rare disease that leads to degeneration of the thalamic nucleus. Main symptoms:
- progressive insomnia;
- autonomic and endocrine dysfunction;
- dysarthria;
- ataxia;
- myoclonus;
- pyramidal symptoms.
Brain dystrophy due to the influence of the prion virus.
Cognitive impairment develops, confusion and hallucinations appear. In the terminal stage, complete insomnia develops. Patients die within 3 years from the onset of the first signs. The disease is associated with the PRNP D178N-129M mutation. There is also a sporadic form of the disorder.
Some information about prions
The researchers note:
- The prion protein includes 254 amino acid residues and weighs 33–35 kilodaltons [2];
- the gene encoding the PrP protein is found in humans, mammals, and birds [1];
- To completely destroy the prion protein, a temperature of at least 1000 degrees is required [1]!
- it is possible that prions take part in intercellular recognition and cellular activation [3];
- it is possible that the function of prions is to suppress age-related processes [3];
- with the development of clinical manifestations of prion diseases there are no signs of inflammation or changes in the blood;
- it is assumed that prions are involved in the development of schizophrenia and myopathy;
- The mechanism of action of prions and their transformation from a normal form to a pathological one remains unclear.
Characteristics of infections
Diseases are caused by pathogens that do not contain nucleic acid. They can be sporadic, genetically determined, infectious (even iatrogenic). The presence of defective proteins causes disorders commonly called spongiform encephalopathies. These are degenerative disorders of the nervous system in which the brain gradually takes on a spongy appearance due to miniature holes due to prions killing neurons.
All infections are incurable and fatal. Today, research and testing are being conducted to explore methods of slowing the progression of prion disease.
Conditions for the occurrence of diseases
The conditions for the occurrence of prion diseases are unique. They can form according to three scenarios: as infectious, sporadic and hereditary lesions. In the latter option, genetic predisposition plays a major role [2].
Renowned prion researcher Stanley Prusiner identifies two striking features of neurodegenerative diseases such as Creutzfeldt-Jakob disease, Alzheimer's disease, and Parkinson's disease. The first is that more than 80% of cases of the disease are sporadic (that is, random, occurring “by themselves”). Second: despite the fact that a large number of mutant proteins specific to a particular disease are expressed during embryonic development, the patterns of inheritance of these neurodegenerative diseases appear later. This suggests that certain processes occur during aging that release disease-causing proteins [5]. More than 20 years ago, the author argued that this process involves the random refolding of a protein into an incorrectly folded one, which corresponds to the transition to an infectious state - a prion.
Interesting facts about Alzheimer's disease: Chronic sleep deprivation may increase the risk of Alzheimer's disease ("New step in understanding Alzheimer's disease: sleep deprivation may be a risk factor"), and Alzheimer's neuropeptide itself (amyloid-β Aβ) may be part of the innate immune system ( "Perhaps Alzheimer's β-amyloid is part of the innate immune system." - Ed.
In the last decade, interest in this topic has renewed due to the possibility of developing diagnostics and effective therapy [5]. Many different explanations have emerged for age-related neurodegenerative diseases, such as oxidative modification of DNA, lipids and/or proteins; somatic mutations; altered innate immunity; exogenous toxins; DNA-RNA mismatches; disruption of chaperone function; absence of one of the gene alleles [5]. An alternative complex explanation is that different groups of proteins can form prions. Although small amounts of prions can be eliminated through protein degradation pathways, excessive accumulation over time allows prions to self-propagate throughout the body (Figure 2), resulting in central nervous system dysfunction [5].
Figure 2. Processes of neurodegeneration caused by prions. Top: The accumulation of a “normal” prion protein increases its likelihood of transitioning to a toxic conformation, which is described by a greater content of β-structure. Prions are most pathogenic in the form of oligomers; After fibril formation, toxicity decreases. Depending on which specific prion protein we are talking about, in a pathological state it can form plaques, tangles or inclusion bodies. Possible routes of drug intervention: (I) reduction of con precursor protein; (II) inhibition of the formation of the prion form; (III) destruction of toxic aggregates. Bottom: Hereditary senile neurodegeneration is explained by two events: the presence of a mutant form of the precursor and the formation of a prion from it, ready for oligo- and polymerization to form toxic forms.
[5]
Prion - neuron killer
A prion, also known in microbiology as infectious PrP Sc, is a professional term describing a defective form of a protein commonly found in mammalian brain tissue that promotes long-term memory function.
Prions kill neurons very quickly, which provokes the rapid development of degenerative diseases.
According to the theory, we are talking about the source of many disorders of the central nervous system of animals (including humans). These proteins play a role in hereditary changes and evolution.
One of the regulatory factors of prion conversion is chaperones, whose function is to clean cells from pathogenic proteins. Both chaperones and prions belong to the group of cellular proteins.
Theories and hypotheses
The theory explaining why prions kill neurons was formulated by Professor S. B. Prusiner in 1982. He was also the first to use the word "prion" (originally intended to be used as a portmanteau of "proteinaceous" and "infectious", but it sounded better in the altered state).
Prusiner formulated the theory of prions as pathogenic proteins in connection with the search for the causative agent of Creutzfeldt-Jakob disorder. The agent he discovered was something new. Until 1982, it was believed that infectious diseases could only be caused by infectious organisms containing nucleic acid carrying genetic information. But the protein structure does not contain nucleic acid. It is a protein that multiplies by changing similar proteins in the body. In 1997, S. B. Prusiner received the Nobel Prize in this field.
Pathogenic prion proteins have the same primary structure (amino acid sequence), but differ in their conformational configuration. While wild PrP c has a strong α-helix predominance and about 5% β-sheet, pathogenic PrP Sc (Sc – scrapie) has a β-sheet proportion of up to 40%. The reasons for the aberrant properties of PrP Sc prions are still under investigation. Today there are several hypotheses.
- The viral hypothesis explains the effects of prions by virology. The involvement of RNA viruses in transmissible spongiform encephalopathies has been suggested. Both viroids and prions are small infectious pathogens; therefore, the result of exposure to the virus is a prion of an infectious nature.
- Multicomponent hypothesis . It is assumed that for the formation of infectious proteins as new pathogens, communication with polyanions and lipids is necessary.
- Heavy metal poisoning . Intoxication causes the development of infection when there is a lack or excess of copper in the body (an optimal amount of copper is necessary for healthy protein).
What is the toxicity of protein to a neuron?
Once the infectious protein appears, it can “imprint” its conformation onto healthy neurons. Toxic proteins can kill mitochondria in neurons, spreading disease to tissues.
Pathogenic proteins are extremely resistant to physical and chemical influences, which leads to difficulties in sterilization (attempts have been made to burn the brains of affected animals at 600°C; the ash subsequently infected approximately 1/3 of the animals). In accordance with what metabolic processes are disrupted by prions in the neuron, the most infectious are the tissues of the eye, brain and spinal cord.
Laboratory diagnosis and treatment
Diagnosis is based on intracerebral infection of mice or hamsters, which slowly (up to 150 days) develop the corresponding disease if the patient was sick [2]. Histological examination of the brains of dead animals is often carried out [2].
Unfortunately, to date, effective methods for treating prion diseases have not yet been developed, although attempts are being made to prevent the conformational transition of a normal protein into an abnormal one. Therefore, the most reliable way to prevent the development of infectious forms is prevention [2].
The solution to the “prion issue” is becoming especially relevant in connection with the growing threat of an epidemic through invasive medical operations and even when taking medications.
Animal diseases
The risk of animal diseases for humans is very speculative (the problem is best characterized by the connection between BSE and a new variant of Creutzfeldt-Jacobs disorder). In animals, disorders are manifested by aggressiveness and impaired motor abilities:
- bovine spongiform encephalopathy (BSE);
- scrapie;
- chronic wasting disease (CWD);
- felt spongiform encephalopathy;
- transmissible encephalopathy.
Prospects
Apparently, interest in prions will not fade until assumptions about them are fully confirmed and effective methods for treating prion diseases are found. The article [6] talks about the need for modern research, which requires careful consideration of foreign prions in extraneuronal tissues.
The authors used mice as model objects: two lines that transgenically expressed sheep prion protein, and one line that expressed human prion protein (Fig. 3). The goal was to compare the effectiveness of interspecies transmission of infection through brain and spleen tissue. Intracerebral infection with a foreign prion protein resulted in the absence or small amount of the infectious agent in the brains of these mice. However, infectious foreign prions were detected in the spleen at earlier stages of infection compared to when neurotropic prions were used, suggesting that lymphatic tissue may be more permissive to the spread of foreign prions compared with the brain.
Figure 3. The ability of the Sc237 hamster prion to infect and transmit when injected into the brain or spleen of transgenic mice having the sheep (tg338; white mice) or human (tg7; gray mice) PrP prion protein. The number of diseased/injected mice is shown in parentheses; Below is the average lifetime (in days).
[6]
What causes this preferential replication of prions in lymphatic tissues is still unknown. However, the findings indicate that humans may be more sensitive to foreign prions than previously thought based on the presence of prions in the brain, and for this reason, asymptomatic carriers of prion disease may not be recognized. This once again confirms that such a powerful biomolecule as the prion is fraught with many mysteries, the disclosure of which may help in understanding a number of insoluble problems of humanity...
Diagnostics
Diagnosis of prion diseases is based on the detection of cognitive disorders, their progressive development, and already at an intermediate stage of development - non-specific EEG disturbances, gradually turning into specific patterns (burst suppression pattern) with myoclonus in the clinical and EMG picture, and epileptic seizures.
Only 14-3-3 protein is present in the cerebrospinal fluid.
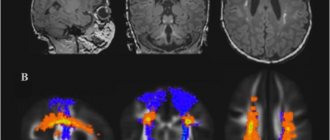
In sporadic Creutzfeldt-Jacobs disease, T2-weighted MRI demonstrates hyperintensity in the nucleus basalis and caudalis. In the new variant of the disease, it is present in the pulvinar thalamus and has the shape of a club.
DNA testing can help identify genetic and familial forms of the disorder, which means identifying the E200K mutation.
The clinical diagnostic conclusion is confirmed by immunohistochemical and neurohistological examination of brain tissue.
Shown:
- typical neurocytic vacuolization;
- spongiomatous degeneration of the neuropil;
- loss of nerve cells;
- astrogliosis;
- presence of scrapie-associated fibrils in the affected brain.
Literature
- Abramova Z.I. Study of proteins and nucleic acids. Kazan: Kazan State University, 2006. - 157 pp.;
- Novikov D.K., Generalov I.I., Danyushchenkova N.M. Medical microbiology. Vitebsk: VSU, 2010. - 597 pp.;
- Prudnikova S.V. Microbiology with basics of virology. Krasnoyarsk: IPK SFU, 2008;
- Pozdeev O.K., Pokrovsky V.I. Medical microbiology. M.: Geotar-med, 2001. - 765 pp.;
- S. B. Prusiner. (2012). A Unifying Role for Prions in Neurodegenerative Diseases. Science
.
336 , 1511-1513; - V. Beringue, L. Herzog, E. Jaumein, F. Reine, P. Sibille, et. al.. (2012). Facilitated Cross-Species Transmission of Prions in Extraneural Tissue. Science
.
335 , 472-475; - Carolina Pola. (2012). Prion escape to spleen. Nat Med
.
18 , 360-360; - Elements: “10 facts about prions and amyloids”;
- Elements: “Geometry of protein bodies”;
- Charles Weissmann. (2012). Mutation and Selection of Prions. PLoS Pathog
.
8 , e1002582.
Taking care of your health
Chinese doctors also reported that patients who have had COVID pneumonia may develop pulmonary fibrosis. What is this, what does it threaten?
Olga Bogush : COVID-19 occurs in different ways. In patients who had a mild or latent, asymptomatic form of illness, consequences most often do not occur. If the patient was diagnosed with bilateral pneumonia caused by coronavirus, especially if more than 25 percent of the lungs were affected, there is a risk of complications, including fibrosis.
What it is? Fibrosis is the replacement of normal lung tissue with connective tissue. The lung essentially becomes scarred. Inextensible areas form in the elastic tissue, and the respiratory surface of the lungs decreases. If two or three such areas appear and they are small, the patient may not notice it. But if the fibrosis is large, breathing problems begin. The most common symptom is shortness of breath. Fibrosis occurs not only under the influence of viral infection. Chronic obstructive pulmonary disease, for example, affects many long-term smokers. In severe cases, this leads to respiratory failure.
Is it possible to prevent the development of such complications?
Olga Bogush : The development of complications depends on many reasons, on the severity of the disease, first of all. But it is possible and necessary to fight to restore lung health. The basis is physical rehabilitation. By the way, the Ministry of Health recently released a large document with recommendations for rehabilitation after COVID-19.
Unfortunately, we do not have special medications or any special technologies to help the lungs function the same way as before the disease. For example, the now popular vitamin D does not protect against COVID-19 and its consequences. Lung tissue must repair itself, and our task is to help it. The main thing a person can do at home is to work on their physical recovery.
First of all, this is breathing exercises. It has features. It is necessary to perform exercises aimed at restoring the respiratory muscles, developing the muscles that are responsible for inhalation and exhalation.
There are many breathing practices. I recommend Alexandra Strelnikova’s gymnastics to my patients; it works very well for almost any chronic lung disease. The recommendations of the Ministry of Health indicate that you can practice yogic breathing. It is interesting that these breathing practices are very different, but both have a good effect on the condition of the lungs.
And, of course, you can limit yourself to only breathing exercises while severe weakness persists. Strelnikova’s gymnastics, by the way, can be performed initially while sitting, and even lying down - it still works.
When severe weakness persists, you can even do very simple things while lying down: for example, inflate balloons or exhale slowly through a thin tube, the end of which is lowered into water. There are special simulators that train you to inhale and exhale - they are beautiful and useful. Although quite expensive. I think it’s quite possible to do without them - just use the means at hand and don’t be lazy. But gradually, having grown stronger, you need to add physical activity.
What exercises do you recommend?
Olga Bogush: Regular gymnastics is already good. If you have an exercise machine at home - a stepper, a walking track, a bicycle - that's great. But even just regular walking, swinging your arms, bending your torso and other basic exercises will help you recover faster.
When the quarantine regime ends and you can go for a walk, you need to add any physical activity in the fresh air. At first, I would recommend aerobic exercise - not intense, but lasting at least half an hour to an hour.
The simplest and safest thing is walking at a brisk pace, or cycling. And intense activity, including strength exercises, can be done later, when the body has recovered from the illness.
What additional recommendations are there?
Olga Bogush : You need to pay attention to your sleep. We see sleep disturbances in patients very often.
Many people are now psychologically wary; they are simply afraid of first getting infected, and then, if they get sick, they are afraid of complications. Stress slows recovery. It is important that nutrition is complete. Increase the amount of protein in your diet - this is necessary for the normal restoration of body tissues.
And I also recommend that all my patients, including older ones, get vaccinated against pneumococcus as a preventative measure. This vaccination will protect against the occurrence of secondary bacterial pneumonia. Vaccines have been known for a long time. They are time-tested. Safe and work very well.
Often, after suffering from bronchitis or pneumonia, a slight cough lasts for a long time - for months, I know from myself. Should he be treated?
Olga Bogush: Residual cough can indeed persist for up to two months. A slight temperature may persist for some time - around 37 degrees, this is not scary.
Sometimes patients themselves delay the process, for example, they use inhalations with mucolytics - lazolvan or some other similar drugs. There is no need to do this. Inhalations for moistening the respiratory tract are useful - with mineral water, saline solution. But if you add mucolytics, it will provoke a cough.
Therefore, if some unpleasant symptoms do not go away over time - shortness of breath increases, your health worsens, the cough takes on a different character, wheezing is added to it, for example - in such cases there is no need to treat yourself.
Are you afraid to go to the doctor at the clinic - now it is possible to get a consultation remotely, there are many telemedicine projects now.
Many, fearing COVID, bought pulse oximeters. After recovery, are they no longer needed or can they come in handy?
Olga Bogush: A pulse oximeter will help monitor how recovery is progressing. The normal level of oxygenation is 95-98 units, not lower. 94 is not enough, this is a reason to consult a doctor. You just need to take into account that there may be temporary fluctuations in indicators. For example, if a person did not sleep well, their oxygenation level will be lower than usual. So you need to focus on your general well-being.
What else helps, besides gymnastics and inhalations?
Olga Bogush : It’s good to perform vibration massage. This is not at all difficult: ask someone to lightly pat your back with your palms for a few minutes, gently and not forcefully “tap” or “slap” your entire back. Helps clear the airways of mucus and works as a preventive measure for congestion in the lungs.
Human prion diseases: symptoms and treatment prospects
The mechanisms of human diseases caused by prions (English from Pr otein (“protein”) and Infect ion (“Infection”; the word was first used by S. Prusiner [1] at the end of the 20th century) still remain poorly understood, despite There seems to be a large amount of research being carried out in this area. The purpose of this article is to summarize and clearly explain the currently available information regarding prions and their associated diseases.
For the purposes of this article, the following abbreviations are accepted: prion - proteinacious infectious particle; PrP - prion protein; PrPC , normal isoform of prion protein; PrPSc —infectious form of prion protein; P RNP is a gene encoding a prion protein.
Prion diseases (also known as transmissible spongiform encephalopathies, TSEs), which became known to man in the mid-18th century, are one of the most intriguing biological phenomena. Research into this phenomenon began in the 20th century with attempts to determine the biological essence of the causative agents of several specific diseases of animals and humans with similar symptoms. The hypothesis about their common etiology, put forward in the 1960s by scientists radiobiologist T. Alper and mathematician D. Griffith [2] and later supplemented and proven by physician S. Prusiner [3], gave impetus to subsequent research in this area. However, despite the deep interest of the scientific world, many aspects of the existence of prions remain unexplored to this day.
Human diseases associated with these specific protein infectious agents include Creutzfeldt-Jakob-Creutzfeldt disease (CJD) and its various variations, fatal insomnia (FFI/FSI), Gerstmann-Schraussler-Scheinker disease (GSS), kuru, variable protease-sensitive prionopathy, and prion disease associated with diarrhea and damage to the autonomic nervous system.
All of the above prion diseases remain fatal today, placing them in the category of the most dangerous diseases.
The essence of prions
After the process of translating genetic information, contained in the form of a nucleotide sequence of RNA (ribonucleic acid), into a specific amino acid sequence that forms the primary structure of all proteins, the newly synthesized proteins fold into certain structures. Prions are a type of misfolded protein molecule (Fig. 1), a defective form of the normal membrane protein PrP, which is expressed (manifests) primarily in cells of the central nervous system.
Rice. 1. Correctly “folded” (left) and defective (right) proteins [4]
Thanks to certain biological machinery, ordinary misfolded proteins are easily utilized and do not have a negative effect on the processes of human life. Prions are distinguished by their resistance to these mechanisms and the ability to convert their normal constituent proteins into similar ones. There are two hypotheses describing the mechanisms of this phenomenon. According to the first, heterodimeric model (Fig. 2) [5], the transformation occurs as follows: PrPSc joins the “healthy” PrP molecule and catalyzes a series of conformational changes leading to its transition to the prion form, after which the two abnormal proteins diverge and launch new rounds of this process. Moreover, the presence of an aggregated (“glued together”) form of the protein is not a necessary part of prion transformation.
Rice. 2. Heterodimeric model of prion replication [6]
An alternative hypothesis (Fig. 3) [5] - polymerization - states that catalysis of the conformational transformation of a normal protein into a pathological one can only occur through “nuclealization” followed by the formation of oligomeric or multimeric complexes. It is worth noting that recent research favors the second model.
Rice. 3. PrPSc replication cycle according to the polymerization hypothesis [7]
Exposure to PrPSc leads to a kind of “intracellular epidemic”: a lot of non-functional protein plaques are formed on the body’s cells, which is why it dies sooner or later.
Pathways of prion diseases
It is believed that there are only three ways of acquiring prion diseases: direct infection, hereditary transmission and sporadic occurrence by an unknown mechanism [8], but regardless of the origin, they can be transmitted by infectious means.
A highly conserved PRNP gene was detected, which carries information about the normal isoform of the PrP protein, located in the p-arm of human chromosome 20 [9]. PRNP has a length of 16 thousand nucleotide sequences and contains 2 exons. All hereditary prion diseases are associated with autosomal inheritance of mutations that occur in this gene.
Rice. 4. Localization of the PRNP gene on human chromosome 20 [10].
The main way prion disease occurs is spontaneous. According to one of the hypotheses explaining this process, a certain post-translational modification occurs in normal proteins [11]. Another hypothesis postulates that at some specific moment an indefinite number of cells in the body somatically (non-hereditarily) mutate and begin to produce the defective PrPSc protein [12].
A prion can enter an uninfected human body in various ways: by eating poorly prepared meat containing PrPSc, by blood transfusion from an infected person to a healthy person, by transplantation of infected organs and tissues.
Clinical picture of human prion diseases
Another important feature of prion proteins is the ability to take on a certain number of different conformations. This causes differences in the course and symptoms of prion diseases: different incubation periods, damage to different parts of the cerebral cortex, and disruption of various functions of the nervous system are possible [13]. Despite this, among all diseases associated with the action of prions, a series of common features can be traced: damage to the nervous system, the initial absence of an immune response to defective PrP proteins due to the constant presence of their “correct” isoform in the body [5], rapid progression of the disease after the end of the incubation period.
Creutzfeldt-Jakob disease ( CJD ) is a rare but best known human prion disease. There are several forms of CJD—hereditary, iatrogenic, sporadic, and variable—with the first three differing primarily in how they spread. The pathological pictures of hereditary, iatrogenic and sporadic CJD are similar: all cases show progressive cognitive impairment, cerebellar lesions and dysfunction, or a combination of these disorders; visual impairment up to blindness; myoclonic seizures. In the terminal stage, global cognitive impairment occurs, and death occurs 8–10 months after diagnosis of CJD [14]. Variable CJD has several deeper distinctive features: it affects young people under the average age of 30 years, its onset is characterized by behavioral changes, insomnia, depression; motor disorders appear approximately 6 months from the onset of the disease in the form of progressive ataxia, chorea, myoclonus; dementia occurs later than in the classic form, the patient is aware of his deteriorating condition. Variable CJD is characterized not only by onset at a younger age, but also by a median survival of more than 14 months [15].
Gerstmann-Straussler-Scheinker disease ( GSS ) is a disease that differs somewhat from CJD in several ways. This clinical form of TSE is caused by a mutation of the PNRP gene at codon 102, leading to the replacement of the amino acid proline with leucine [16]. The disease begins in middle age with the manifestation of cerebellar ataxia, speech disorders, dementia and changes in behavior. These symptoms may include diplopia, deafness, myoclonic seizures, and spasticity.
Kuru is a prion disease endemic to parts of Papua New Guinea. The main way this disease spread was ritual cannibalism. Symptoms include movement disorders (tremor, massive fasciculations, choreoathetosis, myoclonus). Death occurs approximately 2 years after the onset of the disease. In 2009, it was discovered that some members of one of the aboriginal tribes have innate immunity to kuru due to the appearance of a relatively new polymorphic modification of the PNRP gene [17].
Fatal insomnia is a rare TSE usually associated with inheritance of an autosomal dominant mutation (“familial” FFI). It is noted that there is also a sporadic form of this disease (“spontaneous”, FSI) [18]. In both cases, the following picture is observed: sleep disturbances, hallucinations, autonomic hyperactivation, motor disorders, and a sharp and progressive decline in cognitive abilities. The disease lasts from 8 to 72 months (the average is about 18 months), after which the patient dies. There is rapid death of neurons and astrogliosis of the anterior and medial thalamus and inferior olives, with subsequent damage to the cerebral cortex and cerebellum. The stages of the disease are described [18]: the initial form is characterized by the appearance of severe insomnia, panic attacks, pathological anxiety and phobias; at the second stage of the disease, the patient begins to experience hallucinations; at the penultimate stage, the patient loses the ability to sleep and rapidly loses weight; the terminal stage results in loss of speech and death.
In 2013, another clinical form of prion diseases , associated with damage to the autonomic nervous system [19]. The disease is associated with the appearance of a new mutation in the PRNP gene, leading to a shortening of the prion protein and subsequent disruption of its connection with cell membranes. In this case, the distribution of prion aggregates is not limited to the central nervous system. Presumably, PrPSc migrates to peripheral nerves and internal organs. A series of symptoms appear early in life: chronic diarrhea, autonomic failure and sensory polyneuropathy. In adulthood, damage to the central nervous system occurs, leading to the appearance of dementia and seizures among the symptoms.
Variable protease-sensitive prionopathy ( VPSPr ) is another new rare sporadic prion disease, first described in 2008. VPSPr is similar to GSS in terms of PrPSc features, however, no PRNP gene mutations were detected in the VPSPr prion [20]. The immunity of prion proteins that cause VPSPr to the action of proteases is significantly reduced. The disease is manifested by speech disorders (aphasia, dysarthria), cognitive impairment, and in some cases ataxia and parkinsonism.
Prospects for the treatment of prion diseases
Much research in this area provides evidence for the possibility of inhibiting prion replication and treating the diseases they cause.
As noted above, the body's immune response to PrPSc is absent. However, an experiment conducted with in vitro-derived prions proved that the use of antibodies produced against certain antigenic determinants of PrP induces inhibition of PrPSc proliferation [21], leading to a delay in the disease. Prion conversion can also be stopped using “β-structure blockers” - peptide sequences enriched with the amino acid proline and having a composition homologous to PrPC [5]. Another approach is based on the use of antisense oligonucleotides (ASO) - short fragments of nucleic acids that stop translation from messenger RNA due to the formation of loop-like regions on it. It is ASO that is currently the most effective method of inhibiting prion replication: experiments with the introduction of ASO into the cerebrospinal fluid of laboratory mice, conducted at Rocky Mountain Laboratories, led to a delay in the manifestation of prion diseases in experimental subjects by 113–135 days [22] . Astemizole, which belongs to the group of histamine H1 receptor (H1R) blockers, has a proven antiprion effect [23]. In addition, to prevent the multiplication of prions, it is possible to use mutations of the PNRP gene, leading to changes Q171R and E219K in the amino acid sequence of PrP: mutant prion proteins are unable to transform into a pathological form [24].
At the moment, treatment of prion diseases can only be symptomatic. The use of Brefeldin A, which destroys the Golgi apparatus and thereby slows down the proliferation of PrPSc, and NMDA receptor antagonists, which promote longer survival of infected cells, in the treatment of CJD has not achieved much success [25]. Attempts to use classical antiviral agents in the treatment of CJD and GSS also failed [25]. Traditional hypnotics have zero effectiveness in the treatment of FSI and FFI, although a case of delayed death has been reported with the simultaneous use of a number of potent drugs (diazepam, ketamine, nitric oxide) [26]. VPSPr therapy can be based on the use of the increased sensitivity of PrPSc to the action of proteases: delivery to the body of a mixture of specific immobilized cysteine proteases will probably delay the onset of the disease and prolong the life of the patient. Etiotropic treatment of major TSEs, apparently, should be based on the use of the above or similar methods of inhibition of prion replication.
Literature:
- Stanley B. Prusiner - Autobiography. NobelPrize.org.
- Alper T., Cramp WA, Haig DA, Clarke MC Does the agent of scrapie replicate without nucleic acid? //Nature. - 1967. - May (vol. 214, no. 5090). — P. 764–766.
- Taubes, Gary. The game of name is fame. But is it science? // Discover. - 1986. - December (vol. 7, no. 12). — P. 28–41.
- Mayo Foundation of Medical Education and Research.
- I. S. Shkundina, M. D. Ter-Avanesyan. Prions. Advances in biological chemistry, v. 46, 2006. - pp. 7–9.
- Nora Whisler. Lecture 34: PRION. — 2015. — P. 22.
- Dipendra Paj Pandeya, Nimish K. Acharya and Seong-Tshool Hong. Review: The Prion and its Potentiality. // Biomedical Research. — 2010.
- Groschup MH, Kretzschmar HA, eds. Prion Diseases Diagnosis and Pathogeneis // Archives of Virology - New York: Springer, 2001. - Vol. Suppl 16.
- Oesch B., Westaway D., Wälchli M., et al. A cellular gene encodes scrapie PrP 27–30 protein // Cell. - Cell Press, 1985. - Vol. 40, no. 4. - P. 735–746.
- Andreas Papassotiropoulus, Adriano Aguzzi, M. Axel Wollmer, Christoph Hock. The prion gene is associated with human long-term memory. // Human Molecular Genetics. — 2005.
- Prion Diseases (Transmissible Spongiform Encephalopathies) [Online].
- Prion Clinic: Sporadic Prion Disease [Online].
- N. N. Zavadenko, G. Sh. Khondkaryan, R. Ts. Bambeeva, A. A. Kholin, E. N. Saverskaya. Human prion diseases: modern aspects. // Journal of Neurology and Psychiatry. S. S. Korsakova. - 2018. - vol. 118, ed. 6. - pp. 91–92.
- ICTVdB Index of Viruses. US National Institutes of Health website [Online].
- Clinical and Pathologic Characteristics | Variant Creutzfeldt-Jakob Disease, Classic (CJD) [Online].
- Arata H, Takashima H, Hirano R, et al. Early clinical signs and imaging findings in Gerstmann–Sträussler–Scheinker syndrome (Pro102Leu). // Neurology. — 2006.
- Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Campbell T, Al-Dujaily H, Hummerich H, Beck J, Mein CA, Verzilli C, Whittaker J, Alpers MP, Collinge J. A Novel Protective Prion Protein Variant that Colocalizes with Kuru Exposure. // New England Journal of Medicine. — 2009.
- R.Turner. Dying To Sleep: Fatal Familial Insomnia (FFI) [Online].
- Mead S, Gandhi S, Collinge J, Caine D, Gallujipali D, Carswell Ch, Hyare H, Joiner S, Ayling H, Lashley T, Linehan JM, Al-Doujaily H, Sharps B, Revesz T, Sandberg MK, Reilly MM, Koltzenburg M, Forbes A, Rudge P, Brandner S, Warren JD, Wadsworth JDF, Wood NW, Holton JL, Collinge J. A novel prion disease associated with diarrhea and autonomic neuropathy. // New England Journal of Medicine. — 2013.
- Gambetti P, Puoti G, Zou WQ. Variably protease-sensitive prionopathy: a novel disease of the prion protein. // Journal of Molecular Neuroscience. — 2011.
- M. Enari, E. Flechsig, C. Weissmann. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. // Proceedings of the National Academy of Sciences. — 2001.
- G. Raymond, et al. Antisense oligonucleotides extend survival of prion-infected mice. // JCI Insight. — 2021.
- Scripps Research Institute Scientists Identify First Potentially Effective Therapy for Human Prion Disease; Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents // Proceedings of the National Academy of Sciences. — 2013.
- K. Kaneko et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. // Proceedings of the National Academy of Sciences. — 1997.
- M. Maniz, P. Kalakoti, M. Henry, J. Thakur, R. Menger, B. Guthikonda, A. Nanda. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. // Neurosurgical Focus. — 2015.
- David Robson. The tragedy of people who stop sleeping: is it possible to help them? [Online] // BBC Future. — 2021.
Treatment of prion diseases
Prion diseases are currently incurable. Doctors cannot influence the mechanisms of disease development or even slow them down. The life expectancy of a person affected by a prion infection depends on the body’s own reactions.
ATTENTION! The average life expectancy for an adult patient with prion infection is 5 to 7 years.
The only thing doctors can offer is short-term symptomatic therapy. If necessary, anticonvulsants are prescribed (if there are epileptic seizures), hemodialysis for kidney damage due to prion infection.
The mortality rate from prion infection in adults is 100%.