Motor neuron
(from the Latin motor - driving and neuron;
motor neuron
) - a large nerve cell in the anterior horns of the spinal cord. Motor neurons provide motor coordination and maintenance of muscle tone[1][2].
Motor neurons are named by the muscle they innervate (quadriceps, gastrocnemius, semitendinosus, etc.)[2].
There are alpha motor neurons and gamma motor neurons[1][2]:
| View | Function |
| alpha motor neurons | innervate skeletal muscle fibers (extrafusal fibers) and provide muscle contraction |
| gamma motor neurons | innervate stretch receptors (intrafusal fibers) |
Forms of motor neuron disease
Motor neuron diseases are classified according to whether they are inherited or sporadic, and whether the pathology is upper motor neuron or lower motor neuron dependent.
In adults, the most common form of the disease is amyotrophic lateral sclerosis (ALS), which affects both upper and lower motor neurons. The disease has hereditary and sporadic forms and can affect the arms, legs or facial muscles.
Primary lateral sclerosis is an upper motor neuron disease, whereas progressive muscular atrophy affects only the lower motor neurons in the spinal cord.
In progressive bulbar palsy, the lowest motor neurons in the brainstem are most affected, causing symptoms such as slurred speech and difficulty chewing and swallowing.
Symptoms of motor neuron diseases
Below is a brief description of the symptoms of some of the most common forms of motor neurone disease.
Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig's disease or classical motor neuron disease, is a progressive, ultimately fatal disease that disrupts signaling in all muscles of the body. Many doctors use the terms motor neuron disease and ALS interchangeably. The disease is caused by disorders of both upper and lower motor neurons. The first symptoms are usually noticed in the arms and legs or in the muscles responsible for swallowing. About 75 percent of people with classic ALS develop weakness of the bulbar muscles (the muscles that control speech, swallowing and chewing). Muscle weakness and atrophy occur on both sides of the body. Patients lose strength and the ability to move their arms and legs and hold their body upright. Other symptoms include muscle spasticity, spasms, and muscle cramps. Speech may become slurred or guttural. When the muscles of the diaphragm and chest wall do not function properly, patients lose the ability to breathe without mechanical support. Although the disease generally does not impair a person's intellectual abilities or personality, some recent scientific research suggests that some people with ALS may develop cognitive (mental) impairment. Most people with amyotrophic lateral sclerosis die from respiratory failure, usually within 3 to 5 years of the onset of symptoms. However, about 10 percent of patients survive for 10 years or more.
Progressive bulbar palsy , also called progressive bulbar atrophy, affects the lower motor neurons responsible for activities such as swallowing, speaking, chewing, and others. Symptoms include weakness of the glossopharyngeal, jaw and facial muscles, progressive loss of speech function, and atrophy of the tongue muscles. Limb weakness in the disease with signs of motor neuron damage is almost always obvious, but less noticeable. People are at increased risk of choking and aspiration pneumonia caused by the passage of fluids and food through the lower respiratory tract and lungs. Affected individuals have emotional outbursts of laughing or crying (called emotional lability). Stroke and myasthenia gravis may have certain symptoms similar to those of progressive bulbar palsy and should be excluded when diagnosing this disease. In about 25 percent of people with ALS, early symptoms begin with associated bulbar abnormalities. Many clinicians believe that progressive bulbar palsy by itself, without signs of pathology in the extremities (arms or legs), is extremely rare.
Pseudobulbar palsy , which has many symptoms similar to progressive bulbar palsy, is characterized by degeneration of the upper motor neurons that transmit signals to the lower motor neurons in the brain stem. Patients develop progressive loss of the ability to speak, chew, and swallow, as well as progressive weakness of the facial muscles. Patients may develop voice disturbances and an increased gag reflex. The tongue may become immobile and lose the ability to protrude from the mouth.
Primary lateral sclerosis (PLS) damages the upper motor neurons of the arms, legs, and face. This occurs when specific nerve cells in the motor areas of the cerebral cortex (the thin layer of cells covering the brain that is responsible for most high-level brain functions) gradually degenerate, causing movements to be slow. The disease often affects the legs first, then the torso, arms, and finally the bulbar muscles. Speech may become slow and difficult. When nerve cells are damaged, leg and arm movements become clumsy, slow and weak, and spasticity occurs, resulting in the inability to walk or perform tasks that require fine hand coordination. Balance problems can lead to falls. Speech may become slow and slurred. Patients typically experience pseudobulbar affect and an overactive response. Primary lateral sclerosis is more common in men than women, and the onset of the disease usually occurs between 40 and 60 years of age. The cause of the disease is unknown. Symptoms progress gradually over many years, resulting in progressive stiffness (spasticity) and clumsiness in the affected muscles. PLS is sometimes considered a form of amyotrophic lateral sclerosis, but the main difference is the sparing of lower motor neurons, a slow rate of disease progression, and a normal life expectancy. Primary lateral sclerosis may be mistaken for spastic paraplegia, an inherited upper motor neuron disorder that causes spasms in the legs and usually begins in adolescence. Most neurologists follow the clinical course of the affected person for at least 3-4 years before making a diagnosis. This disease is not fatal, but can affect quality of life.
Progressive muscular atrophy is characterized by slow but progressive degeneration of exclusively lower motor neurons. The disease largely affects men, with onset earlier than other motor neuron diseases. Weakness usually begins in the arms and then spreads to the lower body, where it can be more severe. Other symptoms may include muscle wasting, clumsy arm movements, and muscle cramps. The muscles responsible for breathing may be damaged. Exposure to cold may worsen symptoms. The disease develops together with ALS in many cases.
Spinal muscular atrophy (SMA) is an inherited disease affecting the lower motor neurons. This is an autosomal recessive disease caused by disturbances in the SMN1 gene (the gene responsible for the production of a protein that is important for the functioning of motor neurons (SMN protein)). In SMA, insufficient levels of SMN protein lead to degeneration of lower motor neurons, causing weakness and wasting of skeletal muscles. Weakness is often more severe in the muscles of the arms and legs, as well as the muscles of the trunk. Spinal muscular atrophy in children is divided into three types based on age of onset, severity, and progression of symptoms. All three types are caused by defects in the SMN1 gene.
Post-polio syndrome (PPS) is a condition that can affect polio survivors, which can occur for decades after they have recovered from polio. Poliomyelitis is an acute viral disease that destroys motor neurons. Many people affected by the disease early in life recover, only to experience new symptoms many decades later. After acute polio, the surviving motor neurons are responsible for more of the muscles they control. Post-polio syndrome and post-polio muscle atrophy are thought to occur when surviving motor neurons are lost through aging or injury/disease. Many scientists believe that SPP is a hidden symptom of muscle weakness previously affected by polio, rather than a new motor neuron disease. Symptoms include fatigue, slowly progressive muscle weakness, muscle atrophy, cold intolerance, and muscle and joint pain. These symptoms most often occur among the muscle groups affected by the initial disease and may consist of problems breathing, swallowing, or sleeping. Other symptoms of SSP may be caused by skeletal deformities, such as long-standing scoliosis, which have led to chronic changes in the biomechanics of the joints and spine. Symptoms are more common in older people and those most affected by earlier illness. Some people have only minor symptoms, while others develop muscle wasting, which can be misdiagnosed as ALS. SSP usually does not threaten the patient’s life. According to medical statistics, 25 to 50 percent of those who have had paralytic poliomyelitis usually develop post-polio syndrome.
Motor units (MU)
The concept of a motor unit (MU) is given and its structure is described. The classification of MUs and the correspondence of MUs and types of muscle fibers is given. The principle of size and Henneman's rule are described. Data are presented on the activation of motor units during strength exercises depending on the magnitude of the load.
Definition
The term "motor unit" was proposed by E.G. Liddell and C.S. Sherrington to designate a group of muscle fibers innervated by terminals (branches) of one axon.
Currently, a motor unit (MU) is understood as an elementary functional unit of a muscle, which includes a motor neuron and the muscle fibers innervated by it.
DE structure
Having entered the muscle, the motor neuron axon branches into many branches, each of which innervates a separate muscle fiber. Thus, one motor neuron innervates a fairly large number of muscle fibers (from several units to several thousand), while each muscle fiber is innervated by only one motor neuron.
It has been established that muscle fibers belonging to the same motor unit are distributed throughout the muscle, that is, they belong to different muscle bundles.
Such a dispersed (dispersed) distribution of muscle fibers of each MU ensures uniform muscle contraction when only a certain part of the MU is “involved” in the work.
It should be noted that one MU consists of muscle fibers that have the same properties. Through the activation of various motor units, the central nervous system controls the activity of the entire muscle.
MU size (innervation ratio, innervation coefficient)
The size of the motor unit is the number of muscle fibers that are innervated by one motor neuron. To determine this indicator, the number of muscle fibers in skeletal muscle and the number of motor neurons that innervate these muscle fibers are determined (Table 1). Sometimes in the literature the size of the motor unit is called the innervation ratio or innervation coefficient.
Whenever a motor neuron fires, it sends action potentials to all the muscle fibers it innervates.
Therefore, the lower the innervation coefficient , the more perfect the control of the muscle fibers by the nervous system.
Based on the innervation coefficient (MU size), one can judge the number of branches required for the axon of a motor neuron to innervate all the muscle fibers entering the MU.
Table 1 - Number of muscle fibers, number of motor units (motoneurons) and size of motor units in various human skeletal muscles
| Muscle | Number of muscle fibers | Number of units | DU size |
| Anterior tibial | 250090 | 445 | 562 |
| Medial head of the gastrocnemius muscle | 1120365 | 1934 | 579 |
| External rectus oculi muscle | 26730 | 2970 | 9 |
| Brachioradial | 136530 | 333 | 410 |
With age, the number of MUs per muscle decreases. This is due to the fact that the number of motor neurons that innervate an individual muscle decreases. As a result, the number of muscle fibers also decreases as the human body ages.
Classifications DE
There are various classifications of DU. Based on the significance for the body, R. Burke et al. (RE Burke, 1973) proposed dividing motor units according to a combination of two characteristics - contraction speed and resistance to fatigue.
According to this classification, MUs are divided into three types: S (slow) – slow, resistant to fatigue; FR (fast resistant) - fast, resistant to fatigue, FF (fast fatigable) - fast, quickly fatigued.
These motor units correspond to different types of muscle fibers (Table 1).
Table 1 - Correspondence between types of motor units and muscle fibers
| Type DE | S | FR | FF |
| Muscle fiber type | Type I | Type IIA | IIB type |
The structure and functions of a motor neuron correspond to the morphological characteristics of the muscle fibers that it innervates. Thus, the motor neuron DE S type has a small cell body and innervates from 10 to 180 muscle fibers, and the motor neuron DE FF type has a large cell body and innervates from 300 to 800 muscle fibers (J.H. Wilmore, D.L. Costill, 1997 ) (Fig. 1).
Rice. 1. Histochemical and physiological properties of the three main types of motor units and muscle fibers (RE Burke, 1973)
In table Figure 2 shows the number of muscle fibers and the number of MUs in various human muscles
Size principle or Henneman's rule
S-type MUs have a low activation threshold, so when muscle strength develops, they are the first to be activated. After this, FR type MUs are activated. MU FF type have a high activation threshold, therefore, when effort develops in the muscle, they are the last to be activated.
Due to the fact that muscle fibers belonging to different motor units are dispersed throughout the muscle and are not located in one bundle, the development of muscle strength is characterized by smoothness.
However, due to the fact that connective tissue connections exist between neighboring muscle fibers, when some muscle fibers contract, for example, those that are part of the S type MU, and the relaxed state of others (for example, those that are part of the FF type MU), friction forces should arise, causing high muscle viscosity. G.V.
Vasyukov (1967) showed that at low muscle tensions (30% of the maximum), muscle viscosity is maximum. With further tension of the muscle, when many muscle fibers are simultaneously excited, the viscosity of the muscle decreases abruptly.
The structure and functions of muscles are described in more detail in my books “Hypertrophy of Human Skeletal Muscles” and “Muscle Biomechanics”
Diagnosis of motor neuron disease
There are no specific tests to diagnose most motor neuron diseases, although genetic testing for SMA genes currently exists. Symptoms may vary among patients and in the early stages of the disease may be similar to those of other diseases, making diagnosis difficult. The physical examination is followed by a thorough neurological examination. Neurological tests will evaluate motor and sensory skills, nervous system functioning, hearing and speech, vision, coordination and balance, mental status, and changes in mood or behavior.
Tests to rule out other diseases or measure the extent of muscle damage may include the following:
Electromyography (EMG) is used to diagnose lower motor neuron disorders, as well as muscle and peripheral nerve disorders. In an EMG, a doctor inserts a thin needle-shaped electrode attached to a recording instrument into the muscle to evaluate electrical activity during voluntary contraction and at rest. Electrical activity in the muscle is caused by lower motor neurons. When motor neurons are lost, characteristic abnormal electrical signals occur in the muscle. Testing usually takes about an hour or more, depending on the number of muscles and nerves being tested.
EMG is usually performed in combination with nerve conduction velocity testing. Nerve conduction studies measure the speed at which impulses are transmitted in nerves from small electrodes attached to the skin, as well as their strength. A small pulse of electricity (similar to a shock from static electricity) stimulates the nerve that controls a specific muscle. The second set of electrodes transmits a response electrical signal to the recording device. Nerve conduction studies help differentiate lower motor neuron disease from peripheral neuropathy and can detect abnormalities in sensory nerves.
Laboratory tests of blood, urine, and other substances can help rule out muscle diseases and other disorders that may have symptoms similar to those of motor neurone disease. For example, analysis of the cerebrospinal fluid, which surrounds the brain and spinal cord, can detect infections or inflammation that can also cause muscle stiffness. Blood tests may be ordered to measure levels of the protein creatine kinase (necessary for chemical reactions that produce energy for muscle contractions); high levels allow the diagnosis of muscle diseases such as muscular dystrophy.
Magnetic resonance imaging (MRI) uses a powerful magnetic field to produce detailed images of tissue, organs, bones, nerves and other structures of the body. MRI is often used to rule out diseases that affect the head, neck and spinal cord. MRI images can help diagnose brain and spinal cord tumors, eye diseases, inflammation, infection and vascular disorders that can lead to stroke. MRI can also detect and monitor inflammatory diseases such as multiple sclerosis. Magnetic resonance spectroscopy is a type of MRI scan that measures levels of chemicals in the brain and can be used to assess the integrity of upper motor neurons.
A biopsy of muscles or nerves can confirm the presence of neurological disorders, in particular impaired nerve regeneration. A small sample of muscle or nerve tissue is taken under local anesthetic and examined under a microscope. The sample can be removed surgically through an incision in the skin or by biopsy, in which a thin, hollow needle is inserted through the skin into the muscle. A small amount of muscle tissue remains in the hollow needle when it is removed from the body. Although this test can provide valuable information about the extent of damage, it is an invasive procedure and many experts believe that a biopsy is not always necessary for diagnosis.
Transcranial magnetic stimulation was first developed as a diagnostic tool to study areas of the brain associated with motor activity. It is also used as a treatment for certain diseases. This non-invasive procedure creates a magnetic pulse inside the brain that causes movement in an area of the body. Electrodes attached to various areas of the body collect and record electrical activity in the muscles. Measures of evoked activity can diagnose motor neuron dysfunction in motor neuron disease or monitor disease progression.
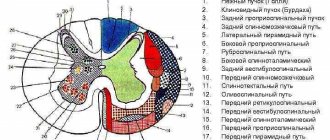
Spinal cord pathways
text_fields
text_fields
arrow_upward
Axons of the spinal ganglia and gray matter of the spinal cord go into its white matter, and then into other structures of the central nervous system, thereby creating the so-called pathways,
functionally divided into
- Propriospinal,
- Spinocerebral,
- Cerebrospinal.
1. Propriospinal tract
connect neurons of the same or different segments of the spinal cord. They start from the neurons of the gray matter of the intermediate zone, go to the white matter of the lateral or ventral funiculi of the spinal cord and end in the gray matter of the intermediate zone or on the motor neurons of the anterior horns of other segments. The function of such connections is associative and consists in coordinating posture, muscle tone, and movements of different metameres of the body. The propriospinal tracts also include commissural fibers that connect functionally homogeneous symmetrical and asymmetrical areas of the spinal cord.
2. Spinocerebral tracts
connect segments of the spinal cord with brain structures.
They are presented
- proprioceptive,
- spinothalamic,
- spinocerebellar,
- spinoreticular pathways.
Proprioceptive pathway
begins from the receptors of deep sensitivity of the muscles of the tendons, periosteum, and joint membranes. Through the spinal ganglion it goes to the dorsal roots of the spinal cord, into the white matter of the posterior cords, and rises to the Gaulle and Burdach nuclei of the medulla oblongata. Here the first switch to a new neuron occurs, then the path goes to the lateral nuclei of the thalamus of the opposite hemisphere of the brain, switches to a new neuron - the second switch. From the thalamus, the pathway ascends to the neurons of the somatosensory cortex. Along the way, the fibers of these tracts give off collaterals in each segment of the spinal cord, which creates the possibility of correcting the posture of the entire body. The speed of excitation along the fibers of this tract reaches 60-100 m/sec.
Spinothalamic tract
starts from pain, temperature,... tactile, skin baroreceptors. The signal from the skin receptors goes to the spinal ganglion, then through the dorsal root to the dorsal horn of the spinal cord (first switching). Sensitive neurons of the dorsal horns send axons to the opposite side of the spinal cord and ascend along the lateral cord to the thalamus (the speed of excitation through them is 1-30 m/s) (second switching), then to the sensory cortex. Some of the fibers of the skin receptors go to the thalamus along the anterior cord of the spinal cord. Somatovisceral afferents also travel along the spinoreticular pathway.
Spinocerebellar tracts
start from the receptors of muscles, ligaments, internal organs and are represented by the non-crossing Govers' bundle and the double-crossing Flexig's bundle. Consequently, all spinocerebellar pathways, starting on the left side of the body, end in the left cerebellum, just as the right cerebellum receives information only from its side of the body. This information comes from Golgi tendon receptors, proprioceptors, pressure and touch receptors. The speed of excitation along these paths reaches 110-120 m/s.
3. Cerebrospinal tracts
start from the neurons of the brain structures and end on the neurons of the spinal cord segments.
These include paths:
- corticospinal
(from pyramidal neurons of the pyramidal and extrapyramidal cortex), which provides regulation of voluntary movements; - rubrospinal,
- vestibulospinal ,
- reticulospinal tract -
regulating muscle tone.
What unites all of these pathways is that their final destination is the motor neurons of the anterior horns
.
Peripheral motor neurons are divided into alpha motor neurons and gamma motor neurons (Fig. 21.2).
Smaller gamma motor neurons innervate intrafusal muscle fibers. Activation of gamma motor neurons increases the stretch of muscle spindles, thereby facilitating tendon and other reflexes through alpha motor neurons.
Each muscle is innervated by several hundred alpha motor neurons. In turn, each alpha motor neuron innervates many muscle fibers - about twenty in the extrinsic muscles of the eye and hundreds in the muscles of the limbs and trunk.
Acetylcholine is released at neuromuscular junctions.
The axons of peripheral motor neurons are part of the cranial nerves and the anterior roots of the spinal cord. At the level of the intervertebral foramina, the anterior roots and posterior roots are acidified, forming the spinal nerves. Several adjacent spinal nerves form a plexus and then branch into peripheral nerves. The latter also branch repeatedly and innervate several muscles. Finally, the axon of each alpha motor neuron forms numerous branches, innervating many muscle fibers.
Each alpha motor neuron receives direct excitatory glutamatergic inputs from cortical motor neurons and from sensory neurons innervating the muscle spindles. Exciting influences also come to alpha and gamma motor neurons from the motor nuclei of the brain stem and interneurons of the spinal cord - both through direct pathways and with switches.
Direct postsynaptic inhibition of alpha motor neurons is carried out by Renshaw cells—glycinergic intercalary neurons. Indirect presynaptic inhibition of alpha motor neurons and indirect presynaptic inhibition of gamma motor neurons are provided by other neurons that form GABAergic synapses on dorsal horn neurons.
Other interneurons of the spinal cord, as well as motor nuclei of the brain stem, also have an inhibitory effect on alpha and gamma motor neurons.
If excitatory inputs predominate, a group of peripheral motor neurons are activated. First, small motor neurons are excited. As the force of muscle contraction increases, the frequency of their discharges increases and large motor neurons are involved. With maximum muscle contraction, the entire corresponding group of motor neurons is excited.