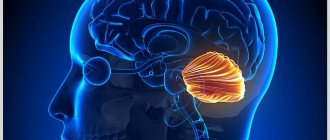
Мозжечковая миндалина — это специфическая часть мозга, визуально напоминающая по форме обычную миндалину. Ее дислокация находится в глубине височной доли головного мозга, на латыни «доля» носит название Lobus temporalis. Каждое полушарие головного мозга обладает своей миндалиной. Значение этих миндалин сложно переоценить, ведь они участвуют в формировании эмоций, являются составным элементом лимбической структуры.
У людей и многих видов животных именно данная часть головного мозга ответственна за вырабатывание позитивных и негативных эмоций – удовольствие, страх, гнев и другие. Размер мозжечковой миндалины обладает прямой зависимостью относительно агрессивных действий. Данная часть мозга сексуально-диморфна. К примеру, если мужчин был подвержен кастрации, то мозжечковые миндалины уменьшаются в размере на 30%.
Медиками было доказано, что многие психические заболевания связаны именно с нарушением нормальной функции мозжечковых миндалин. В частности, такие заболевания:
- повышенная тревожность;
- аутизм;
- шизофрения;
- биполярное расстройство;
- различные фобии.
Причины появления
Дистопия мозжечка, как правило, является врожденной патологией. Она возникает при смещении какого-либо органа в эмбриональный период. Вторичной она бывает лишь при проведении частых пункций или при люмбальных травмах. Иных причин появления данного заболевания не выявлено.
Миндалины мозжечка очень сходны с теми, которые находятся в гортани. В нормальном положении они располагаются выше БЗО черепа. А отклонения в их развитии и положении могут повлечь за собой не только дистопию. Чаще всего встречается опущение миндалин мозжечка ниже уровня черепа.
До сих пор заболевание Киари является патологией, о причинах возникновения которого неврологи не пришли к одному мнению. Некоторые придерживаются мнения, что эта аномалия возникает при уменьшении габаритов ямки позади черепного выходного отверстия к спинному мозговому каналу. Это нередко приводит к таким последствиям в процессе роста тканей, которые расположены в коробке. Они выходят в затылочный выходной канал. Иные специалисты полагают, что заболевание начинает развиваться по причине увеличения объемов мозговых тканей головы. В этом случае мозг начинает выталкивать через заднюю черепную ямку в затылочное черепное отверстие мозжечок и его миндалины.
Синдром Арнольда-Киари
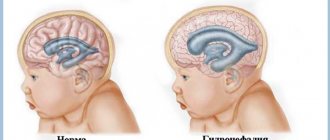
Низкое расположение миндалин мозжечка – врожденное состояние, происходит опущение структур мозга головы в затылочное отверстие. В процесс, как правило, вовлекаются продолговатый мозг и мозжечок. Выявляется болезнь Арнольда Киари у некоторых людей совершенно случайно, к примеру, при обследовании из-за другой патологии. На первой стадии течение заболевание легкое и часто незаметное. В этой ситуации мальформация Арнольда-Киари не представляет угрозы для здоровья организма.
Протекать патология может практически бессимптомной, но часто этот болезнь сочетается сирингомиелией (название заболевания серого вещества спинного мозга). Отсутствие лечения может спровоцировать гидроцефалию (скопление в черепе жидкости), инфаркту мозга и прочим опасным патологиям, отмечены случаи инвалидности. Возможность выявить порок можно сразу после рождения или через 20-30 лет. Аномалия Киари может быть одного из 4 типов.
Аномалия Арнольда Киари 1 степени – структуры заднего отдела нередко ущемляются из-за выхода через затылочное отверстие. Приводит такое положение к скоплению спинномозговой жидкости. Синдром Киари первой степени представляет собой смещение миндалин мозжечка, располагаются они ниже затылочного отверстия. Часто обнаруживается аномалия Арнольда-Киари в подростковом возрасте.
Синдром Арнольда-Киари 2 степени
Аномалия второй степени имеет более выраженные структурные изменения. Вовлекаются в процесс мозжечок, которые располагается в большом затылочном отверстии. Скрининговые УЗИ плода могут показать некоторые пороки строение позвоночника, спинного мозга. Прогноз в большинстве случаев для жизни малыша благоприятен, но клинические признаки патологии будут присутствовать. Это указывает на необходимость динамического наблюдения за пациентом.
В этом случае практически все образования черепной ямки располагаются ниже большого затылочного отверстия (мост, 4 желудочек, продолговатый мозг, мозжечок). Часто они находятся в шейно-затылочной мозговой грыже (когда присутствует дефект позвоночного канала, при котором дужки позвонков незаращены, содержимое дурального мешка, в который входят все оболочки и спинной мозг). При таком типе аномалии большое затылочное отверстие имеет увеличенный диаметр.
Это самый тяжелый вариант заболевания. При данном варианте синдрома Арнольда Киари наблюдается гипоплазия мозжечка, недоразвитие. Часто сочетается с врожденными кистами задней черепной ямки, врожденными кистами и гидроцефалией. При диагностировании 4-го типа прогноз неблагоприятный, в большинстве случаев болезнь заканчивается смертью больного.
Типы аномалий
Есть такие виды аномальных отклонений – дистопия и аномалия Киари.
В свою очередь, заболевание Киари разделяют на четыре различных типа:
- Тип I отличается расположением миндалин ниже уровня большого затылочного отверстия. Определяется такая патология, как правило, у подростков и у взрослых. Часто ее сопровождает скопление спинальной ликворной жидкости в центральном канале, где расположен спинной мозг пациента.
- Тип II характеризуется проявлением сразу после появления на свет. Кроме того, помимо миндалин, при втором типе патологии в отверстие затылочной части выходят червь мозжечка с частью продолговатого мозга и желудочек. Второй тип аномалии много чаще сопровождает гидромиелия, чем при патологии, описанной в первом случае. В большей части случаев такое патологическое отклонение связано с врожденными грыжами, образованными в различных отделах спинного мозга.
- III тип отличают опустившиеся сквозь отверстие не только миндалины, а и мозжечок вместе с тканями продолговатого мозга. Они размещаются в шейном и затылочном отделах.
- Тип IV представляет собой недоразвитие тканей мозжечка. Эта патология не сопровождается смещением их в каудальном направлении. Но при этом аномалию чаще всего сопровождает врожденная киста, располагающаяся в черепной ямке, и гидроцефалия.
II и III типы часто проявляются в сочетании с явлениями дисплазии нервной системы, например, с гетеротопией мозговых тканей коры, кистами отверстия и пр.
Симптомы
Самой распространенной среди аномалий является патология первого вида. При ней нередко возможно проявление ликворно-гипертензионного синдрома, а также церебелло-бульбарное и сирингомиелическое явления, нарушения работы нервных окончаний внутри черепа.
Ликворно-гипертензионный синдром – это боли в затылке и шейных мышцах, которые усиливаются во время чихания, кашля или напряжения шейных мышечных тканей. Часто боли сопровождаются рвотой, не связанной с приемами еды. Многие симптомы патологии проявляются в зависимости от того, в каком положении располагаются миндалины мозжечка относительно отверстия, которое находится в затылочной ямке черепа. Наблюдается также:
- повышенный тонус мышц шейного отдела;
- нарушения речевых функций;
- ухудшение работы органов зрения и слуха;
- отклонения при глотании;
- частые головокружения, сопровождаемые шумом в голове;
- ощущение вращения окружающей обстановки;
- непродолжительные обмороки;
- перепады давления при резких движениях;
- атрофия языка;
- сиплость голоса;
- нарушения дыхания и чувствительности разных частей тела;
- приступы онемения;
- нарушения в тазовых органах;
- ослабление мышц конечностей.
Аномалия II и III типов имеет похожие симптомы, заметные уже с первых мгновений после рождения малыша. Второй тип сопровождает шумное дыхание, а также неожиданные приступы остановки дыхания, нейропарез гортанных тканей. Наблюдаются также отклонения процесса глотания.
Признаки дистопии редко бывают очевидными. Но все же возможны неврологические проявления:
- приступы «стреляющих» болей в шейных мышцах при усилении напряжения или кашле;
- частые боли в области головы;
- приступы головокружений и обмороки.
Если опущение миндалин сильное, иногда наблюдается расширение канала, связывающего головной и спинной мозг, а также образовываются полости вокруг канала.
Диагностические методы
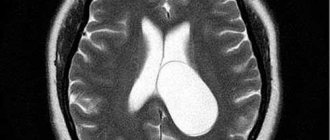
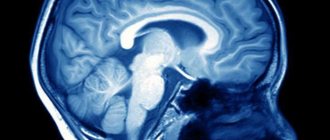
Основным современным методом диагностирования дистопии является МРТ. В этом случае ни КТ, ни рентгеновские исследования не дают полной картины патологии.
Для диагностирования синдрома Киари не подойдут никакие стандартные методы типа ЭЭГ, ЭхоЭГ или РЭГ, потому как они не позволяют точно поставить диагноз. Осмотр невролога тоже не определит аномалию. Все эти методы могут показать подозрение только на повышенное давление внутри черепной коробки. Рентгенографию черепа также не стоит делать, поскольку она показывает лишь аномалии костных тканей, которые могут сопровождать патологию. Потому до введения в диагностическую практику томографии диагностировать эту болезнь было проблематично. Современные же способы диагностирования позволяют точно определить патологию.
В случае качественной визуализации костных тканей вертебрального перехода такие методы, как МСКТ или КТ, не дают достаточно точной картины. Единственным достоверным способом диагностирования аномалии Киари на сегодня является только МРТ.
Поскольку проведение исследования этим методом требует неподвижности больного, маленьких детей погружают в искусственный сон с помощью лекарств. Проводится также МРТ спинного мозга. Она направлена на диагностику любых аномальных отклонений в работе нервной системы.
Лечение
Консервативные методы лечения возможны лишь при очень незначительных отклонениях. Все зависит от того, каково состояние больного на момент обращения к врачу. В этом случае лечение направлено на снятие болезненных симптомов нестероидными лекарствами или миорелаксантами. Необходима также коррекция режима.
Единственным эффективным методом лечения при обширных отклонениях является хирургическое вмешательство, которое заключается в расширении ямки черепа и пластики твердых мозговых тканей оболочки.
Показаниями для оперативного лечения являются:
- изнуряющие головные боли, которые не снимаются препаратами.
- нарастание мозговых проявлений, приводящее к инвалидности.
Если аномальное отклонение протекает без каких-то ощутимых признаков, лечения не требуется. В случаях возникновения болезненных ощущений в районе шеи и затылка проводится консервативная терапия, при которой используют анальгетики и асептические лекарственные вещества, а также миорелаксанты.
При сопровождении аномалии Киари нарушениями неврологических функций или когда консервативный курс терапии не дает результатов, назначается хирургическая операция.
Часто при лечебных курсах синдрома Киари используется метод краниовертебральной декомпрессии. Операция подразумевает расширение отверстия затылочной части за счет удаления части костной ткани, отсечения мозжечковых миндалин и части двух позвонков шеи. Благодаря этому, нормализуется оборот цереброспинальной жидкости в мозговых тканях в результате выполнения заплаты из аллотрансплантата или искусственного материала. Иногда синдром Киари лечат с помощью шунтирования, которое позволяет дренировать цереброспинальную жидкость из центрального канала. Посредством хирургической операции можно отвести цереброспинальную жидкость в сосуды органов груди или брюшины.
Прогноз
Аномалия Киари первого типа может протекать бессимптомно всю жизнь. И третий тип патологии почти всегда приводит к смертельному исходу, если не осуществить своевременное лечение. В случае появления неврологических признаков заболевания первого или последнего типов очень важно своевременно проведенное хирургическое лечение, потому как появившийся недостаток неврологических функций будет плохо восстанавливаться, даже если успешно провести манипуляции. По разным данным, результативность хирургической операции отмечается примерно в половине эпизодов.
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4–6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Какие функции выполняют миндалины?
Благодаря мозжечковым телам, человек способен быстро и надолго запоминать эмоциональные реакции на разного рода события. А так как миндалевидное тело является частью лимбической структуры, человек обладает возможностью бессознательного обучения. Данной особенностью отличаются некоторые типы животных.
Головной мозг имеет особое строение, по этой причине происшествия, значимые в плане выживания, фиксируются при помощи сильной эмоции. Ведь основная работа мозга – это забота о выживании. Соответственно, такая эмоция активизирует внутренней механизм, необходимый для того, чтоб человек не забыл данное событие. Данная информация занимает расположение в долговременной памяти.
Бессознательное обучение может управлять формированием условных рефлексов. Данное обучение выполняется автоматически и на бессознательном уровне. Так как человеческие рефлексы располагаются в мозговых частях, которые не зависят от мышления, их сложно рационализировать. При исполнении своих обязанностей, миндалевидное тело взаимодействует с гиппокампом и базальными ганглиями. Вследствие этого взаимодействия, поступающие данные усваиваются на более высоком уровне.
Дистопия миндалин мозжечка – это специфическое опущение мозжечкового миндалевидного тела в крупное затылочное отверстие. Данную патологию могут именовать мальформацией Киари, происходит когда имеет место каудальная дислокация миндалевидного тела правой либо левой части головного мозга. Для болезни характерно пониженное стояние миндалевидного тела.
Подобное стояние миндалин никоим образом не отражается на жизнедеятельности больного и не вызывает беспокойств. У ребенка данную болезнь находят крайне редко, ей больше подвержены взрослые в промежутке от 30 до 40 лет. Диагностируется случайно, при плановых обследованиях либо лечении других проблем.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Сопутствующие заболевания
Сирингомиелия шейного отдела позвоночника встречается в ~35% (варьирует от 20 до 56%), гидроцефалиия в 30% [1,3] случаев, в обоих случаях считается данные изменения развиваются в результате нарушения ликвородинамики, центральном канале и вокруг спинного мозга.В ~35% (23-45%) выявляются скелетные аномалии [1, 3]:
- платибазия / базилярная импрессия
- атланто-затылочная ассимиляция
- деформация Шпренгеля (Sprengel)
- синдром Клиппель-Фейля (Klippel-Feil)
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Рентгенологические особенности
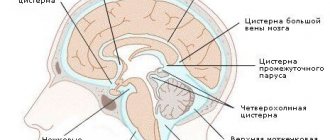
Патология выявляется путем измерения максимального расстояния на которое миндалины выступают ниже плоскости большого затылочного отверстия (условной линии между ophisthion и basion), значения используемые для постановки диагноза отличаются у разных авторов [2]:
- выше затылочного отверстия: норма
- от 3 до 6 мм: неопределенные данные, необходимо сопоставить с симптоматикой, наличием сирингомиелии и т.д.
- {amp}gt; 6 мм: аномалия Арнольда Киари 1
Некоторые авторы используют более простую градацию [1]:
- выше затылочного отверстия: норма
- {amp}gt; 5 мм: аномалия Арнольда Киари 1
Положение миндалин мозжечка меняется с возрастом. У новорожденных миндалины расположены чуть ниже большого затылочного отверстия и спускаются ниже с ростом ребенка, достигая своей низшей точки в возрасте 5 – 15 лет. В дальнейшем они поднимаются на уровень большого затылочного отверстия [3]. Таким образом, снижение миндалин на 5 мм у ребенка будет скорее всего нормой, а взрослом возрасте данные изменения следует рассматривать с подозрением [3].