Генетическое заболевание Верднига-Гофмана относится к группе спинальных амиотрофий, наследуется по аутосомно-рецессивному типу.
Спинальная мышечная атрофия (СМА) характеризуется врожденными или приобретенными дегенеративными изменениями в поперечнополосатых мышцах, симметричной мышечной слабостью туловища, конечностей, отсутствием или снижением сухожильных рефлексов при сохранении чувствительности.
Морфологические исследования выявляют патологию двигательных нейронов спинного мозга, «пучковую атрофию» в скелетных мышцах с характерным чередованием пораженных волокон и здоровых.
Отмечается нарушение проводящей функции нервных волокон, снижение сократительной способности мышц. Статистика
1 из 40-50 человек является носителем мутантного гена SMN. Проявляется патология с частотой 1 : 6 000 — 10 000 новорожденных.
Причины заболевания
Основной причиной спинальной амиотрофии Верднига Гоффмана является мутация гена SMN (от англ. survival motor neuron). Располагается ген выживания мотонейрона на 5 хромосоме, представлен двумя копиями:
- SMNt — теломерная копия, функционально активная;
- SMNc — центромерная копия гена, частично активная.
Продуктом этого гена является белок SMN, участвующий в образовании и регенерации РНК.
Нехватка белка вызывает патологии двигательного нейрона.
В 95% случаев болезни Верднига-Гофмана наблюдается делеция (выпадение) SMNt, что вызывает дефицит белка SMN. Копия SMNc лишь частично компенсирует отсутствие теломерной копии.
Количество копий SMNc составляет от 1 до 5. Чем больше число центромерных копий, тем полнее воспроизводится белок и менее выражена патология нейрона.
Кроме количества копий SMNc, тяжесть заболевания определяется длиной участка делеции и генными конверсиями еще 3 генов: NAIP, H4F5, GTF2H2. Участием дополнительных модифицирующих факторов объясняется клиническое разнообразие симптомов.
Виды и симптомы
Заболевание спинальная амиотрофия разделяется медиками и учеными на несколько типов, отличающихся степенью прогрессирования патологического течения, возрастом пациента, тяжестью течения болезни и ее клиническими признаками, а также продолжительностью жизни.
При этом следует отметить, что в клинической картине всегда отсутствует нарушения, кусающие чувствительной и умственной сферы, равно как и не затрагиваются функции органов таза. Спинальная мышечная атрофия затрагивает только двигательную сферу, именно этим разнится клиническая картина каждого вида патологии.
Что касается принципа деления, медики выделяют 4 формы заболевания, для полноценного понимания, стоит рассмотреть каждую отдельно.
1 форма
Этот тип патологического процесса часто называют младенческим, так как он диагностируется у новорожденных с первых дней жизни и вплоть до 6 месяцев. Первая форма считается наиболее злокачественной в силу возраста человека, а также скорости прогрессирования и наносимого организму ущерба.
Касательно клинической картины, она разнообразна:
- Нарушаются глотательный и сосательный рефлексы, что сопровождается неполноценной моторикой языка. На языке заметны спонтанные и неконтролируемые мышечные сокращения, их называют фисцикуляциями, орган атрофирован. Ввиду нарушений глотательного рефлекса при кормлении молоко матери может попадать в дыхательные пути.
- В этом возрасте характерным симптомом будет дыхательная недостаточности, вызванная нарушением сократительных функций диафрагмальной и межреберной мышц. Отставание развития моторики, ребенок неспособен самостоятельно переворачиваться, удерживать голову, отсутствует хватательный рефлекс, в дальнейшем малыш не сможет сидеть.
- Часто при первой форме патологического процесса происходит деформация костей грудной клетки, она становится ассиметричной, может быть впалой или выпирать.
Развитие патологии с первых дней жизни приводит к тому, что большинство детей не доживают до 6 месяцев.
2 форма
Еще одна форма раннего проявления спинальной амиотрофии, которую принято считать промежуточной. До полугода у ребенка не выявляется никаких признаков болезни, патологический процесс диагностируется в промежутке между шестым месяцем и вторым годом жизни малыша.
Для второй формы развития заболевания характерно постепенное нарастание симптомов:
- В первую очередь утрачиваются рефлексы сухожилий.
- Ухудшается процесс ходьбы, если к моменту начала болезни он уже научился ходить, вскоре навык утрачивается полностью.
- По мере прогрессирования заболевание атрофия мышечной ткани охватывает все конечности.
- Вскоре ребенок перестает держать голову, замечается тремор конечностей.
- Часто в этом случае патология сопровождается тяжелыми формами костно-суставных аномалий, сколиозом позвоночника, вывихами коленных и тазобедренных суставов и т.д.
И при первой, и при второй форме болезни развивается дыхательная недостаточность. Однако прогноз жизни в данном случае более благоприятный. При соответствующем лечении дети преодолевают подростковый возраст, в зависимости от ситуации доживают до 20-30 лет.
3 форма
Этот тип заболевания также имеет название спинальная амиотрофия Кугельберга-Веландера, диагностируется у детей более старшего возраста в промежутке между вторым и пятнадцатым годами жизни.
Клиническая картина в этом случае такова:
- Первый признак – нарушение ходьбы. Ребенок часто спотыкается, виляет, шаги становятся неустойчивыми, виной всему все та же атрофия мышц, в дальнейшем навык утрачивается.
- Как и в прошлом случае через какое-то время болезнь охватывает и верхние конечности.
- Поражаются мышцы лица, затрагивается мимика, чего при прошлых формах патологии не наблюдается.
- Вновь наблюдаются различные деформации скелета.
При патологии спинной атрофии третьей формы, учитывая поддержку медикаментозной терапией, пациенты доживают до 40 лет в среднем.
4 форма
Бульбоспинальная амиотрофия Кеннеди или же взрослая форма заболевания, которая начинается уже после 35 лет жизни при наличии в организме ранее упомянутого поврежденного хромосома.
По статистике этот тип синдрома наиболее благоприятный, если так вообще можно говорить. Патология развивается медленно, но по мере прогрессирования проявляются такие клинические признаки болезни:
- Снижение рефлекторных функций.
- Мышечная слабость в конечностях.
- Масштабная мышечная атрофия на разных участках тела.
- К концу жизни может быть выявлено полное снижение двигательных функций.
- Примечательно, что дыхательный процесс в данном случае не нарушается.
Жизнь пациентов с четвертой формой патологического процесса столь же продолжительна, как и у здоровых людей.
Симптоматика заболевания
СМА 1 и СМА 2 имеет разные симптомы и признаки.
Какие существуют отдаленные последствия черепно мозговой травмы и как максимально себя обезопасить от получения повреждения головы.
Разрыв вен и сосудов головного мозга провоцирует такое заболевание, как субдуральная гематома головного мозга. В чем сложность лечения и диагностики заболевания.
Форма спинальной амиотрофии Верднига СМА 1
Первые симптомы выявляют еще во время беременности по слабому шевелению плода.
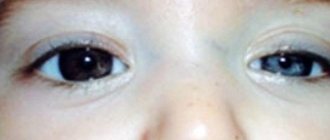
Фото: спинальная амиотрофия Верднига Гофмана
С самого рождения у детей наблюдается дыхательная недостаточность, врожденная спинальная амиотрофия Верднига Гофмана отмечаются:
- низкий мышечный тонус, ребенок не держит голову, не может перевернуться;
- отсутствие рефлексов;
- нарушения сосания, глотания, подергивание языка, пальцев, слабый плач.
Статья в тему: Бронхиальная астма у детей – симптомы и лечение, профилактика заболевания
Малыш принимает характерную позу «лягушки» с согнутыми в суставах руками и ногами, лежа на животе. При СМА 1 нередко отмечают частичный паралич диафрагмы – синдром Кофферата.
Явление характеризуется затруднением дыхания, одышкой, цианозом.
На стороне паралича наблюдается выбухание грудной клетки, повышается риск пневмонии.
У младенцев наблюдаются деформации костной системы, выражающиеся в ограничении подвижности суставов, появлении сколиоза, изменении формы грудной клетки.
Форма СМА 2
Первые месяцы жизни дети развиваются нормально: вовремя начинают держать головку, сидеть, стоять.
После 6 месяцев появляются первые симптомы, обычно после острой респираторной или пищевой инфекции.
В первую очередь страдают конечности, особенно ноги, снижаются сухожильные рефлексы.
Затем в процесс постепенно вовлекаются мышцы туловища и рук, межреберные мышцы, диафрагма, что вызывает деформацию грудной клетки. Изменяется походка, приобретая сходство с «заводной куклой».
Дети становятся неловкими, часто падают. Наблюдаются подергивания языка, дрожание пальцев.
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана
— это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время — дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Клиническая картина заболевания
Спинальная амиотрофия имеет несколько форм и разновидностей
, каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности
, поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.
Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна
, поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
Классификация заболевания
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов :
- Врожденная форма патологии
. Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, и 4 степени сколиоза . Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу; - Ранняя детская форма.
При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет; - Амиотрофия Кугельберга-Веландера.
Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
Диагностика
При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При обнаружении делеции теломерной копии SMNt диагноз считают подтвержденным.
В случае отсутствия делеции проводят дополнительные исследования:
- электонейромиографию;
- исследование нервной проводимости;
- тест на креатинкиназу;
- биопсию мышц и нервной ткани.
При нормальных показателях фермента креатинкиназы проводят подсчет копий SMNc. В случае единственной копии идентифицируют точечную мутацию, принимая окончательное решение.
Дифференциальная диагностика
Похожие симптомы наблюдаются при врожденной миопатии – нарушении тонуса мышц.
Полностью исключить мышечную гипотонию позволяют результаты биопсии.
Определенное сходство с заболеванием Верднига-Гофмана имеет острый полиомиелит. Он начинается бурно, с резкого подъема температуры, несимметричных множественных параличей.
Несколько дней длится острый период, затем процесс переходит в восстановительную стадию.
Гликогенозы и врожденные миопатии также характеризуются сниженным мышечным тонусом. Изменения вызываются, в отличие от спинальной мышечной амиотрофии, нарушением обмена веществ, карциномой, гормональным дисбалансом. Следует исключить также болезнь Гоше, синдром Дауна, ботулизм.
Помогающие организации
Благотворительный фонд «Семьи СМА» — единственная в России организация, специализирующаяся на помощи семьям, столкнувшимся со спинальной мышечной атрофией. Оказывается благотворительную, информационную, психологическую поддержку семьям, консультирует специалистов по вопросам заболевания и методов работы с пациентами со СМА.
Фонд помощи хосписам «Вера». Благотворительная и консультативная помощь семьям с неизлечимо больными детьми, неизлечимо больным взрослым.
ОДКБ № 1 Отделение паллиативной помощи детям, Екатеринбург, Свердловская область. Оказывается медицинскую, информационную, социальную и психологическую помощь семьям, воспитывающим ребенка-инвалида с паллиативным состоянием.
“Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева” ФГБОУ ВО РНИМУ им.Н.И.Пирогова. Институт находится в Москве. Обращаться за медицинской помощью детям со СМА и другими нервно-мышечными заболеваниями могут жители всей России.
Клиника «Чайка». Консультации врача-пульмонолога Штабницкого Василия Андреевича для детей и взрослых со СМА.
Детский хоспис «Дом с маяком» (Москва, ближнее Подмосковье). Медицинская, психологическая, правовая, социальная, благотворительная помощь семьям с неизлечимо больными детьми и молодыми взрослыми (до 25 лет).
Марфо-Мариинский медицинский (г.Москва). Медицинская, психологическая, правовая помощь, помощь няни, мероприятия, духовная поддержка, благотворительная помощь семьям с неизлечимо больными детьми.
Более подробную информацию вы найдете на сайте благотворительного фонда “Семьи СМА” и их специального проекта о жизни со спинальной мышечной атрофией
Спецпроект предназначен тем, кто столкнулся с диагнозом СМА и хотел бы узнать все самое важное об этой болезни: к каким специалистам и куда обращаться, как ухаживать, за чем следить, о чем помнить, какая терапия существует на сегодняшний день.
К изучению Советы по лечению смещения позвонков поясничного отдела
Лечебные методики
Лечение спинальной амиотрофии симптоматическое и направлено на стабилизацию состояния пациента.
Назначают лекарственные средства:
- улучшающие метаболизм – церебролизин, липоцеребин, аминалон;
- влияющие на трофику мышечной ткани – оротат калия, глютаминовая кислота, метионин, токоферола ацетат;
- способствующие нервно-мышечной проводимости – прозерин, галантамин, дибазол;
- стимулирующие кровообращение в капиллярах – компламин, никотиновая кислота;
- поддерживающие жизнеспособность двигательных нейронов – вальпроевую кислоту, рилузол, L-карнитин.
Статья в тему: Таблетки от тахикардии и сердцебиения
Больным предписывают ортопедические процедуры в сочетании с теплыми ваннами, показаны лечебная гимнастика, мягкий массаж, оксигенотерапия, сульфидные ванны.
Виды спинальных амиотрофий
Условно различают проксимальные и дистальные формы СМА. 80% всех видов спинальных амиотрофий относятся к проксимальной форме.
К ним относятся, кроме заболевания Верднига-Гофмана:
- СМА 3 или болезнь Кульдберга-Веландер — заболевают в возрасте от 2 лет до 20, первыми страдают мышцы таза. Отмечается тремор кистей, лордоз.
- Летальная X-сцепленная форма — описана в 1994 году Baumbach, наследуется по рецессивному признаку, наблюдаются преимущественно поражения мышц таза и плечевого пояса.
- Инфантильная дегенерация — нарушаются рефлексы сосания, глотания, дыхание. Смерть может последовать в возрасте до 5 месяцев.
- СПА Рюкю — ген сцепливания не выявлен, наблюдается отсутствие рефлексов, мышечная слабость конечностей после рождения.
В эту группу входит также болезнь Нормана, СМА с врожденным артрогрипозом, СМА с врожденными переломами.
К дистальным спинальным амиотрофиям относится прогрессирующий детский паралич Фацио-Лонде, болезнь Брауна-Виалетта-ван Лэре, СМА с параличом диафрагмы, эпилепсией и глазодвигательными нарушениями.
Как ставится диагноз
Чтобы выявить болезнь внутриутробно, делают забор амниотической жидкости
Выявить спинальную мышечную атрофию Верднига-Гоффмана можно до или после рождения ребенка. Пренатальная диагностика основана на выявлении генетического дефекта. Для этого требуется получить генетический материал ребенка из пуповинной крови, амниотической жидкости или ворсин хориона. Инвазивные методы диагностики опасны для развития беременности, поэтому родители часто отказываются от них. Если патология выявлена, это показание к аборту.
Рождение детей со спинальной мышечной атрофией (СМА) Верднига-Гоффмана возможно по нескольким причинам. Не во всех больницах есть возможность дородовой диагностики болезни и генетического консультирования родителей до зачатия малыша. Родители проводят анализ в основном по собственному желанию и могут до момента родов не знать о том, что малыш будет тяжело болен. Иногда диагноз ставится на позднем сроке, когда прерывать беременность поздно. Играет роль и принципиальное неприятие абортов у некоторых родителей – даже зная о будущей патологии, они принимают решение рожать.
После родов диагноз ставит неонатолог или детский невролог. Важен неврологический статус маленького пациента – нарушение двигательной активности и угасание рефлексов при сохранении чувствительности. Для постановки окончательного диагноза назначается анализ ДНК. МРТ и КТ позвоночника важны, чтобы отличить СМА от других патологий. Нарушения в двигательных ядрах спинного мозга не визуализируются.