Причины
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц. Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна. При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Профилактика и прогноз
Супругам, в роду которых имелись случаи наследственных заболеваний, перед планированием беременности необходимо посетить врача-генетика. Профилактика патологии также заключается в проведении пренатальной диагностики. Выявив миопатию на ранних сроках, можно прервать беременность.
Миопатия Дюшена — наследственная патология, отличающаяся тяжелым течением и быстрым прогрессированием. Это современная медицинская проблема, характеризующаяся разрушением мышечной ткани и быстрым развитием мышечной слабости. Все без исключения больные погибают в раннем возрасте из-за развития не совместимых с жизнью осложнений. Только адекватная и комплексная терапия, четкое соблюдение рекомендаций врача, тщательный уход и забота родителей могут замедлить ход болезни.
Больные быстро становятся инвалидами и погибают в совсем юные годы. Самое страшное, что детям с миопатией не в силах помочь даже квалифицированные врачи, современные медицинские технологии и терапевтические методики 21 века. Болезнь до сих пор остается неизлечимой, забирая молодые жизни. Современные ученые-медики всего мира трудятся над созданием радикального способа борьбы с миопатией Дюшенна.
Симптомы
Начало развития симптомов начинается в раннем детском возрасте, чаще от 1 до 3 лет. Изначально отмечается отставание в моторном развитии, ребенок поздно начинает ходить, часто спотыкается при ходьбе, быстро устает. Позже развивается постоянная патологическая утомляемость мышц. Ребенок практически не может взбираться по лестнице. Походка начинает напоминать «утиную».
Характерным симптомом является симптом «лесенки»: попытка встать из положения сидя происходит с использованием рук, постепенно, медленно, в несколько этапов.
Постепенно начинает отмечаться атрофия мышц, вначале проксимальных отделов нижних, потом верхних конечностей. Позже атрофируются мышцы тазового пояса, бедер, спины, плечевого пояса. Почти всегда развивается «осиная» талия, искривление позвоночника, выпирание лопаток (крыловидные лопатки).
Практически всегда имеет место характерный симптом прогрессирующей мышечной дистрофии Дюшенна – псевдогипертрофия мышц голеней. Мышцы, хоть и увеличены в объеме, они не имеют достаточной силы, очень болезненны на ощупь.
Можно выделить три стадии заболевания: — I стадия – слабость проявляется лишь при значимой физической нагрузке (обычно первый год течения болезни). — II стадия – затруднен подъем по лестнице, быстро развивается слабость при ходьбе. — III стадия – представляет собой параличи, контрактуры мышц с невозможностью cсамостоятельного передвижения.
По видам течения подразделяется на: Быстрое прогрессирование. Способность к передвижению утрачивается быстро, в течение первых 4-5 лет с начала заболевания. Средний темп прогрессирования: передвигаться пациент не может спустя 10 лет. Медленное прогрессирование: выраженных двигательных нарушений через 10 лет от начала болезни нет. Обычно такой вариант характерен для других типов мышечных дистрофий, нежели дистрофии Дюшенна.
ФАКТЫ О МЫШЕЧНОЙ ДИСТРОФИИ ДЮШЕННА/БЕККЕРА
ЧТО ТАКОЕ – МЫШЕЧНАЯ ДИСТРОФИЯ ДЮШЕННА/БЕККЕРА? Мышечные дистрофии – это генетические заболевания, характеризующиеся прогрессирующим истощением и слабостью мышц, начинающихся с микроскопических изменений в них. По мере того, как мышцы разрушаются – их сила уменьшается.
На ранних стадиях МДД и МДБ поражаются мышцы груди (сдвигающие плечи), туловища, а также верхние и нижние мышцы ног. Слабость этих мышц вызывает трудности во вставании, подъёме по ступенькам и удержании равновесия..
Мышечная дистрофия Дюшенна (МДД) впервые была описана в 1860 году французским неврологом Guillaume Benjamin Amand Duchenne. Мышечная дистрофия Беккера (МДБ) названа пo имени немецкого доктора Peter Emil Becker, описавшего этот вариант МДД в 1950 году.
При МДД признаки мышечной слабости обычно проявляются у мальчиков в возрасте около 3 лет. Болезнь постепенно ослабляет скелетные или произвольные мышцы рук, ног и туловища. Примерно в раннем подростковом возрасте или даже раньше могут быть затронуты также сердце и дыхательные мышцы.
МДБ – более мягкая форма МДД. Ее начало обычно приходится на подростковый возраст или раннюю юность, и протекает она медленнее и намного менее предсказуемо, чем МДД.
(Хотя МДД и МДБ болеют почти исключительно мальчики, в редких случаях этими заболеваниями могут болеть и девочки. См. «Это семейное?»)
ЧЕМ ВЫЗВАНЫ МЫШЕЧНЫЕ ДИСТРОФИИ ДЮШЕННА И БЕККЕРА?
До 1980 года было очень мало известно о причинах любых типов мышечных дистрофий. В 1986 году исследователи идентифицировали ген, дефекты в котором, называемые мутациями, вызывали МДД. В 1987 году ассоциированный с этим геном протеин был идентифицирован и назван дистрофином
Гены содержат в себе коды, или рецепты, для протеинов (белков), являющихся очень важными биологическими компонентами всех форм жизни. МДД развивается тогда, когда определенный ген, расположенный в Х-хромосоме, теряет способность вырабатывать протеин дистрофин. МДБ вызвана несколько иными мутациями в том же самом гене. У людей с МДБ присутствует некоторое количество дистрофина, однако его либо недостаточно, либо ухудшено его качество. Наличие некоторого количества дистрофина при МДБ предохраняет мышцы от столь же тяжелой и быстрой дегенерации, как при МДД.
Мышцы состоят из пучков волокон (клеток). Группа независимых белков, расположенных вдоль мембраны, окружающей каждое волокно, помогают нормально функционировать мышечным клеткам.
Когда один из этих белков, дистрофин, отсутствует – это вызывает мышечную дистрофию Дюшенна, если же его недостаточно, или он неполноценный – развивается мышечная дистрофия Беккера
Между прочим, употребление или неупотребление пищи, богатой протеинами, не может возместить утерянный дистрофин. Подробнее о том, как мутация гена приводит к развитию дистрофий Дюшенна и Беккера – см. «Это семейное?»
ЧТО ПРОИСХОДИТ С МЫШЦАМИ ЛЮДЕЙ С ДИСТРОФИЯМИ ДЮШЕННА И БЕККЕРА?
Миодистрофия Дюшенна
Развитие МДД довольно предсказуемо. Дети с этим заболеванием часто поздно начинают ходить. В этот период родители могут заметить увеличение икроножных мышц, или гипертрофию. В дошкольном возрасте дети с МДД могут выглядеть неловкими и часто падать. Вскоре появляются проблемы с подъёмом по лестнице, вставанием с пола или бегом.
В школьном возрасте дети могут начать ходить на пальцах или подушечках пальцев ног с немного перекатывающейся походкой. Походка становится переваливающейся и неустойчивой, и они легко могут споткнуться и упасть. Пытаясь удержать равновесие, они выпячивают живот и откидывают назад плечи. Также возникают трудности с подъёмом рук.
Почти все дети с МДД теряют способность к хождению в период от 7 до 12 лет. В юношеском возрасте для активности рук, ног и туловища требуется помощь или механические приспособления.
Миодистрофия Беккера
Часто диагноз «мышечная дистрофия Беккера» нельзя поставить до подросткового возраста или даже ранней юности, например, когда молодые люди замечают, что у них возникают трудности с занятиями физкультурой или военной подготовкой. Пытаясь скомпенсировать слабость мышц, мальчики начинают ходить переваливающейся походкой, на пальцах ног или подушечках пальцев, выпячивая живот.
Как и при МДД, истощение мышц при МДБ обычно начинается с бедренной и тазовой области, бедер и плеч. Однако при МДБ степень мышечной дегенерации отличается у разных людей в широких пределах. Одним требуется инвалидное кресло к возрасту 30 лет или чуть позже, в то время как другие долгие годы обходятся минимальными приспособлениями, такими, как трость.
КАКИЕ ТЕСТЫ ИСПОЛЬЗУЮТСЯ ДЛЯ ДИАГНОСТИКИ МДД/МДБ?
В диагностике любых форм мышечной дистрофии доктор обычно начинает со знакомства с пациентом и семейной историей и проведения медосмотра. Из этого можно почерпнуть довольно много, включая характер слабости. С истории и осмотра начинается долгий путь к предстоящему установлению диагноза еще до того, как будут проведены любые более сложные диагностические тесты.
Поскольку ослаблены мышцы ног, мальчики с МДД применяют характерный способ вставания с пола, называемый приемом Говерса. Вначале они опираются на руки и колени, затем поднимают таз, а затем «шагают» руками вверх по ногам, чтобы поднять все тело.
Важно получить правильный диагноз, поскольку у других заболеваний некоторые симптомы совпадают с таковыми при МДД/Б. МДБ зачастую может быть невыявлена, либо ошибочно диагностирована как конечно-поясная мышечная дистрофия (КПМД) или спинальная мышечная атрофия (СМА). По этой причине важно провести как генетическое исследование, так и мышечную биопсию до того, как вынести решение, что это именно МДБ.
Доктор также хочет определить, вызвана ли слабость мышц проблемами в них самих, либо в нервах, осуществляющих контроль этими мышцами. Нервы, контролирующие мышцы, или мотонейроны, исходящие из спинного и головного мозга и протягивающиеся ко всем мышцам, могут вызывать мышечную слабость, похожую на ту, что вызвана проблемами в мышцах, хотя в действительности это разные вещи.
Обычно природа слабости может быть уточнена при медосмотре. Иногда проводится специальное исследование, называемое электромиография или исследование нервной проводимости. В процессе этого исследования измеряется электрическая активность мышц и стимулируются нервы, чтобы увидеть, лежит ли проблема в мышцах или нервах.
На раннем этапе диагностики доктора часто назначают исследование крови для определения уровня КФК. КФК обозначает креатинфосфокиназа, энзим, просачивающийся из поврежденных мышц. Когда уровень КФК в крови повышен, это обычно означает, что мышцы разрушаются в результате какого-то патологического процесса, такого как мышечная дистрофия или воспаление. Следовательно, высокий уровень КФК наводит на мысль, что мышечная слабость вызвана патологическими процессами в самих мышцах, но не может точно указать, какое именно это может быть мышечное заболевание.
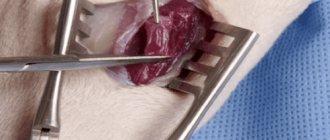
Чтобы определить, какое заболевание является причиной проблем, доктор может назначить мышечную биопсию, хирургическое удаление небольшого кусочка мышцы у пациента. Исследуя этот образец, доктор может много сказать о том, что же в действительности происходит с мышцами. Современные методики позволяют на основании биопсии отличить мышечные дистрофии от воспалительных и иных заболеваний, а так же отличать различные формы дистрофий
Другие тесты с использованием биоптатов могут дать информацию о том, какой протеин присутствует в мышечных клетках, и присутствует ли он в нормальном количестве и на своем ли месте. Это может помочь отличить МДД (дистрофин отсутствует) и МДБ (наличие некоторого количества неполноценного дистрофина). Также может быть назначен МР (магнитный резонанс). Это безболезненное сканирование позволяет доктору визуально определить, что происходит внутри ослабленной мышцы.
Доступность диагностических исследований ДНК, в которых используются либо клетки крови, либо мышечные клетки для получения точной генетической информации, быстро развивается. Вы можете уточнить у своего врача или генетического консультанта, какие тесты доступны. Поскольку многие мужчины с МДБ (и некоторые с МДД) становятся отцами, важно точно знать, какое именно наследственное заболевание у человека. Сестры людей с МДД или МДБ также могут пройти тест, чтобы выяснить, являются ли они носительницами заболевания, поскольку в этом случае у них могут быть дети с этим заболеванием.
ЭТО СЕМЕЙНОЕ?
Узнав о том, что у ребенка – генетическое заболевание, такое как МДД или МДБ, смущенные родители часто спрашивают: «Но в нашей семье этого не было, как же это может быть генетическим?»
МДД может быть характерной для семьи, даже если она есть только у одного члена семьи. Это обусловлено механизмом наследования генетических заболеваний.
И МДД, и МДБ наследуются по так называемому Х-сцепленному типу. Это означает, что ген, мутация в котором и вызывает заболевание, расположен на Х-хромосоме
Каждый ребенок мужского пола получает от матери Х хромосому, а от отца – Y хромосому, которая и делает его мальчиком. Дети женского пола получают две Х хромосомы, по одной от каждого родителя.
Каждый сын, рожденный женщиной с мутацией в гене дистрофина в одной из двух ее Х-хромосом, имеет 50% вероятность унаследовать поврежденный ген и иметь МДД или МДБ. Каждая дочь такой женщины имеет 50% вероятность унаследовать мутацию и стать носительницей. У носительниц обычно нет симптомов заболевания, но у них могут быть дети с мутацией или заболеванием.
Так как же в семье, в которой не было случаев МДД или МДБ, вдруг рождается сын с этим заболеванием?
Этому может быть два объяснения:
Генетическая мутация, приводящая к развитию МДД или МДБ, может присутствовать у женщин на протяжении нескольких поколений, и никто не будет об этом знать. Возможно, не рождались мальчики с заболеванием, или, если даже мальчик в ранних поколениях был болен, родственники могли не знать, что это была за болезнь.
Другое объяснение – это то, что ребенок с МДД или МДБ имеет новую генетическую мутацию, возникшую в процессе его внутриутробного развития. Как только у кого нибудь появляется генетическое заболевание, даже если мутация – спонтанная (новая) у этого человека, он может передать его своему потомству.
Мужчины с МДД или МДБ не могут передать поврежденный ген своим сыновьям, поскольку передают им Y-хромосому, а не Х. Но они безусловно могут передать его своим дочерям, поскольку каждая дочь получает от отца только Х-хромосому. Они будут носительницами, и у каждого их сына будет 50% вероятность того, что у них будет это заболевание, и так далее.
Хороший способ узнать больше о типе наследования в вашей семье – поговорить со своим врачом или геноконсультантом. Можно также посмотреть брошюру .
ЖЕНЩИНЫ И МДД
Почему у девочек не бывает МДД или МДБ? Когда девочка наследует поврежденный ген от матери, обычно она также получает «здоровый» ген дистрофина от отца, вырабатывающий достаточно протеина для того, чтобы предотвратить развитие заболевание. Мальчики, наследующие мутантный ген – болеют, поскольку у них отсутствует второй ген дистрофина, компенсирующий повреждение первого.
Тем не менее, хотя у девочек обычно МДД или МДБ не проявляется в полном объеме, некоторые женщины – носительницы поврежденного гена все же до некоторой степени больны. Небольшой процент женщин – носительниц мутации – так называемые «выраженные носители», и болезнь проявляется у них в мягкой форме.
У таких женщин дефицит дистрофина может проявляться слабостью мышц спины, рук и ног и их быстрой утомляемостью. У выраженных носителей также есть проблемы с сердцем, которые могут выражаться одышкой или неспособностью выполнять простые упражнения. Проблемы с сердцем, если их не лечить, могут быть довольно серьезными и даже опасными для жизни.
Всем женщинам – потенциальным носительницам МДД/МДБ было бы разумно пройти полное диагностические обследование для выяснения своего статуса. В дальнейшем, если подтвердится носительство, регулярная оценка силы и наблюдения за сердцем могут помочь справиться с симптомами, склонными к ухудшению.
ЧТО МОЖЕТ БЫТЬ СДЕЛАНО ДЛЯ ЛЕЧЕНИЯ МДД/МДБ?
Благодаря достижениям во многих областях медицины существуют очень хорошие терапевтические методы, способные помочь при всех проявлениях мышечных дистрофий Дюшенна и Беккера. Эти методы воздействия постоянно совершенствуются. Используя все доступные методы, пациенты могут продлить себе бодрость, активность и продолжительность жизни.
Контрактуры
Влияние болезни можно существенно минимизировать, сохраняя тело насколько возможно гибким, прямым и подвижным. Существует несколько способов добиться этого.
По мере разрушения мышц у больных с мышечной дистрофией часто развивается малоподвижность суставов, называемая контрактурами. Если ими не заниматься, они станут достаточно серьезно выраженными, вызывая дискомфорт и ограничивая подвижность и гибкость. Контрактуры могут затронуть колени, бедра, ступни, локти, запястья и пальцы.
Тем не менее, существует много способов минимизировать и отсрочить контрактуры. Упражнения на амплитуду движений, выполняемые регулярно, помогают замедлить контрактуры посредством предохранения сухожилий от преждевременного укорочения. Очень важно, чтобы физиотерапевт показал вам, как правильно выполняются эти упражнения.
Шины на руках и голенях также могут помочь сохранить конечности растянутыми и подвижными, задерживая начало развития контрактур.
Когда контрактуры сформируются, ослабить их может помочь хирургическое вмешательство. Процедура удлинения сухожилия, называемая операцией на ахилловом сухожилии, часто проводится для лечения контрактур в щиколотке, когда ребенок еще сохраняет способность к хождению. Обычно ребенку требуется после этого использование шин на ногах.
Искривления позвоночника
У подростков с МДД позвоночник может постепенно принять искривленную форму. Это искривление может произойти из стороны в сторону (сколиоз), либо в продольном направлении с принятием формы горба (кифоз). Иногда у тех, кто еще ходит, наблюдается вогнутое искривление в поясничном отделе позвоночника, называемое лордозом.
Сильный сколиоз может мешать сидению, сну и даже дыханию, поэтому желательно его не допускать.
О том, какие упражнения необходимы для сохранения ровной спины, насколько это возможно, а также о правильных позициях во время сидения и сна, можно проконсультироваться с физиотерапевтом
Хирургический метод исправления искривления заключается во введении в позвоночник металлических стержней. Обычно такие операции проводятся в возрасте 11-13 лет
Медикаменты
Медикаментами являются группа препаратов, известных как кортикостероиды, продемонстрировавших эффективность в замедлении прогрессирования МДД (данных «за» или «против» кортикостероидов при МДБ недостаточно)
В 2005 году American Academy of Neurology опубликовала рекомендации по применению этих препаратов при МДД. Они заключаются в следующем:
•Преднизолон или дефлазакорт имеют эффект в терапии МДД. Семилетние исследования показали, что их применение увеличивает силу и улучшает временнЫе характеристики мышц (таких как например время, затраченное на подъем по ступенькам) а также функцию легких
•Эффективные начальные дозы: 0,75 мг на кг массы тела ежедневно для преднизолона, и 0,9 мг на кг массы тела – для дефлазакорта
•Доза должна быть снижена при наличии серьезных побочных эффектов, таких как значительное увеличение массы тела, истончение костей (остеопороз), а также поведенческие проблемы. Наиболее частые побочные эффекты – увеличение массы тела и формирование округлого одутловатого лица
•Пока нет четкой уверенности, что применение дефлазакорта имеет меньшие побочные эффекты, чем преднизолона
Оптимальный возраст начала кортикостероидной терапии не определен. Некоторые врачи уверены, что ее необходимо начинать сразу после постановки диагноза, в то врема как другие предпочитают дождаться времени, когда у мальчиков возникают первые проблемы с хождением. До начала кортикостероидной терапии врач должен обсудить с родителями ожидаемые положительные и потенциальные побочные эффекты
В сочетании с преднизолоном часто назначаются кальциевые добавки и витамин D для нейтрализации его нежелательного воздействия на кости
Обычно рекомендуется низкокалорийная диета с малым содержанием натрия для регулирования массы тела и наблюдаемой при приеме кортикостероидов задержке жидкости.
Иногда при МДД или МДБ назначаются препараты для снижения нагрузки на сердце (см. «Как еще МДД и МДБ воздействуют на организм?»)
Фиксаторы, вертикализаторы и кресла-коляски
Фиксаторы, также называемые ортезами, поддерживают голень и стопу, или охватывают колено. Голеностопные ортезы иногда назначают для применения ночью, чтобы предотвратить отвисание стопы ребенка во время сна
Стояние в течение некоторого времени в течение дня, даже с минимальной весовой нагрузкой, содействует лучшей циркуляции, укреплению костей и выпрямлению позвоночника. Помочь в этом людям с МДД и МДБ могут ходунки или вертикализаторы. Некоторые кресла-коляски также имеют вертикальное положение
Рано или поздно всем мальчикам с МДД требуется кресло-коляска. Многие используют вначале кресла-коляски в школе или на прогулке, продолжая ходить дома. При МДД необходимость постоянного использования коляски наступает как правило в возрасте около 12 лет. Хотя многие дети и их родители воспринимают кресла-коляски как символ инвалидности, большинство считает, что их использование позволяет быть более мобильным, активным и независимым, нежели при попытках ходить во что бы то ни стало на очень слабых ногах.
Другие приспособления могут помочь тем, кто ухаживает за людьми с МДД или МДБ. Среди самых простых – пересадочные площадки для помощи в пересадке с коляски или на коляску. Также можно использовать механические (чаще – гидравлические) подъемники, складные стулья и кровати с электронным управлением.
КАК ЕЩЕ МДД И МДБ ВОЗДЕЙСТВУЮТ НА ОРГАНИЗМ?
Боль и чувствительность
Вы можете быть спокойны, зная, что истощение мышц при МДД и МДБ обычно безболезненно само по себе. Некоторые люди говорят о периодических мышечных спазмах, которые обычно снимаются безрецептурными болеутоляющими средствами
Также, поскольку мышечная дистрофия не затрагивает напрямую нервы, люди с этим заболеванием сохраняют нормальное осязание и другие чувства. Они также как правило контролируют гладкие, или непроизвольные мышцы мочевого пузыря и кишечника, и сохраняют нормальную сексуальную функцию.
Сердце
Аналогично мышцам конечностей, сердечная мышца также может быть ослаблена вследствие недостатка дистрофина. С течением времени, иногда еще до достижения возраста 10 лет, кардиологические проблемы в связи с МДД могут стать жизнеугрожающими. Таким образом, неоходимо тщательное наблюдение за сердечно-сосудистой системой, осуществляемое как правило детским кардиологом.
Вследствие дефицита дистрофина у людей с МДД и МДБ часто развивается кардиомиопатия – слабость сердечной мышцы. Мышечный слой сердца (миокард) вырождается так же, как и скелетные мышцы, что может привести к фатальным кардиологическим проблемам
У некоторых людей с МДБ не так серьезно выражено поражение скелетных мышц, но при этом – серьезные кардиологические проблемы
В 2005 American Academy of Pediatrics сформулировала рекомендации для людей с МДД и МДБ, а также носителей этих заболеваний.
Для пациентов с МДД рекомендовано проходить полное кардиологическое обследование в раннем детстве, и в дальнейшем – раз в 2 года до достижения возраста 10 лет. В дальнейшем обследование необходимо проводить каждый год, или при появлении симптомов слабости сердца, таких как задержка жидкости и одышка.
Для пациентов с МДБ рекомендовано обследование по крайней мере раз в два года, начиная с возраста 10 лет
У носителей МДД и МДБ риск развития кардиомиопатии выше среднего. Специалисты полагают, что носителям необходимо проходить полное кардиологические обследование в поздней юности или ранней зрелости, или скорее – когда проявляются симптомы, и в дальнейшем – проходить такие обследования каждые 5 лет, начиная с 25-30 летнего возраста
Есть предварительные данные, что лечение с применением ангиотензин-превращающих энзимов (АПФ) и бета-блокаторов может замедлить повреждение сердечной мышцы при МДД и МДБ, если лечение начать, как только обнаруживаются отклонения на эхокардиограмме (ультразвуковое исследование сердца), не дожидаясь появления симптомов
Некоторые пациенты с МДБ с серьезными кардиологическими проблемами на фоне неплохого общего состояния были успешно пролечены методом трансплантации сердца.
Дыхательная функция
Когда мальчики с МДД достигают возраста около 10 лет, диафрагма и другие мышцы, управляющие работой легких, ослабевают, и легкие уже менее эффективно выполняют свою функцию. Проблемы, сигнализирующие о недостаточной дыхательной функции – это головные боли, ослабление мыслительной активности, трудности с концентрацией или сохранением бодрости, кошмарные сновидения.
Люди с ослабленной дыхательной системой также более подвержены инфекциям и испытывают трудности при кашле. Простое переохлаждение может привести к развитию пневмонии. При развитии инфекции очень важно получить немедленную медицинскую помощь, дабы не допустить тяжелой дыхательной недостаточности
Когда дыхательная функция ослабевает, семья может приобрести аппарат вентиляции легких, либо научиться процедурам, способствующим откашливанию и сохранению бронхов свободными от выделений. Получить необходимую информацию можно у терапевта или пульмонолога
В некоторых случаях может потребоваться принудительная вентиляция для обеспечения достаточного движения воздуха в легкие и из легких. Иногда дыхательная маска требуется только ночью. Если это необходимо более часто, возможно проведение трахеотомии (для обеспечения поступления воздуха в легкие непосредственно в трахею вставляется трубка)
Существуют эффективные неинвазивные системы вентиляция, позволяющие не использовать трубки. Даже тем, у кого установлены трубки в трахее, иногда возможно отсоединять их от аппарата на некоторое время в течение дня. Более современные трубки имеют в конструкции клапаны, позволяющие говорить.
Интеллектуальные способности
Около трети мальчиков с МДД имеют некоторую степень отставания познавательных способностей, а некоторые – весьма серьезное. Специалисты полагают, что дефицит дистрофина в головном мозге может вызвать познавательные и поведенческие отклонения. Проблемы обучения, наблюдаемые у некоторых людей с МДД и МДБ, проявляются в трех основных областях: фокусировка внимания, словесное обучение и память, и эмоциональное взаимодействие.
Если есть подозрение, что у ребенка некоторая задержка в умственном развитии, необходимо обратиться к детскому нейропсихологу. Если это подтверждается, необходимо незамедлительно начинать обучающее и психологическое вмешательство. Специалист может порекомендовать упражнения и другие способы для взаимодействия с ребенком, которые помогут ему восполнить этот дефицит.
МОГУТ ЛИ СПЕЦИАЛЬНЫЕ ДИЕТЫ ИЛИ УПРАЖНЕНИЯ ПОМОЧЬ ПРИ МДД И МДБ?
Диета
Многие люди, услышав слова «утрата белка» логично задают вопрос: «Мне нужно потреблять больше белка?» К сожалению, потребление пищи с большим содержанием белка не оказывает никакого эффекта на белки, отсутствующие при мышечной дистрофии
Неизвестно ни о каких специальных диетических ограничениях или дополнениях, способных помочь при МДД или МДБ. Комбинация малоподвижности и слабости мышц живота может вызывать тяжелые запоры, поэтому диета должна быть с высоким содержанием жидкости и клетчатки, с преобладанием свежих фруктов и овощей.
У мальчиков, пользующихся электрическими колясками, принимающими преднизолон и не слишком активных вероятно необходимо ограничивать калорийность пищи для поддержания веса. Ожирение дает дополнительную нагрузку на и без того ослабленные мышцы и сердце. Специалисты считают, что низкокалорийная диета не оказывает никакого вредного воздействия на мышцы.
Для принимающих преднизолон и имеющих проблемы с сердцем также может понадобиться диета с низким содержанием натрия.
Упражнения
Упражнения могут помочь в формировании мышц, поддержании здоровой сердечнососудистой системы и способствовать хорошему самочувствию. Однако при мышечной дистрофии излишние упражнения могут повредить мышцы. Проконсультируйтесь с врачом о наилучшей физической нагрузке. При МДД и МДБ возможны умеренные физические нагрузки, но не до изнеможения.
Некоторые эксперты рекомендуют плавание и упражнения в воде (акватерапию) как хороший способ подержания тонуса мышц, насколько это возможно, не вызывая их избыточного перенапряжения.
Поддерживающая способность воды может помочь предупредить некоторые виды напряжения и повреждения мышц. До начала любой программы физических упражнений необходимо пройти кардиологическое обследование.
Физио- и трудотерапия
Физиотерапия обычно является частью комплексной терапии при МДД и МДБ. Для оценки физического состояния и разработки программы физиотерапии необходимо обратиться к физиотерапевту. Основные цели физиотерапии – сохранение подвижности суставов, предупреждение контрактур и сколиоза.
Трудотерапия более сфокусирована на специфической активности и функциях, в отличие от физиотерапии, акцентированной на мобильности, и, где возможно, усилении больших мышечных групп.
Трудотерапия может помочь в решении задач, связанных с работой, отдыхом и повседневной жизнью, таких как передвижение, одевание или пользование компьютером.
КАК С ЭТИМ ЖИТЬ?
Когда у одного из членов семьи МДД или МДБ, вся семья испытывает потребность в поддержке и эмоциональные реакции. Многие черпают помощь и поддержку из религиозных источников, общения с семьями с аналогичным опытом, книг по психологии или консультаций со специалистами. Такие специалисты обычно рекомендуют следующее:
Для детей
•Отвечайте на вопросы детей о болезни по мере их взросления честно и доступным языком
•Всегда представляйте ребенка как личность, с болезнью лишь как одним из аспектов его жизни
•Акцентируйтесь на том, что ребенок может сделать, и помогайте ему делать то, что он хочет. Дети часто находят способы для занятий спортом или другими увлечениями
•Воспитывайте его, как любого другого ребенка, запасаясь терпением, ответственностью, надеждой и любовью. Избегайте избыточной его опеки и помогайте стать независимым
•Предпринимайте нормальную семейную активность, включая отпуска и развлечения. Проявив воображение и терпение, вы можете найти способы делать почти все
Для семьи
•Будьте внимательны к эмоциям и уровню стресса друг друга, проявляйте терпение и доброжелательность
•Планируйте регулярный отдых от обязанностей по заботе
•Решайте проблемы, связанные с болезнью, по мере их поступления. Не фокусируйтесь на будущих осложнениях
•Дайте себе кредит по затрачиваемым усилиям и трудности своих обязанностей
•Организуйте команду поддержки и просите о помощи, когда это необходимо
Научные исследования для поиска методов лечения МДД
С 1986 года, когда был идентифицирован ген, мутации в котором вызывают МДД и МДБ, ученые далеко продвинулись в понимании механизмов этих заболеваний. В настоящее время разрабатываются несколько направлений в поиске способов того, как остановить или обратить разрушение мышц при этих заболеваниях
Одни исследователи создали работающий ген дистрофина без мутаций, и в настоящее время тестируют его безопасность в небольшом клиническом исследовании с участием мальчиков с МДД
Другие исследователи тестируют PTC124, препарат, изменяющий способ «чтения» клетками генетической информации. Примерно у 15% пациентов с МДД молекулярный стоп-сигнал расположен слишком рано для того, чтобы мог синтезироваться полноценный дистрофин. PTC124 заставляет клетки игнорировать этот сигнал
Еще одни исследователи экспериментируют с антисмысловыми нуклеотидами, составами, разработанными, чтобы побудить клетки обойти некоторые типы генетических ошибок, а не только стоп-сигнал. Эти составы прошли лабораторные тесты, и первые клинические испытания продемонстрировали многообещающие результаты
Другие группы исследователей используют стволовые клетки, выделенные из мышц, кровеносных сосудов или костного мозга в попытках добиться регенерации мышц
И, наконец, несколько групп разрабатывают стратегии усиления синтеза протеина атрофин, который очень похож на дистрофин, но синтезируется у людей с МДД и МДБ. Эксперименты показывают, что повышение уровней атрофина может до некоторой степени компенсировать дефицит дистрофина
Краткий обзор основных исследований можно прочесть
По материалам
Диагностика
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. [!] В сложных диагностических ситуациях проводят цитологическое исследование.
Осложнения
Мышечная дистрофия Дюшенна сокращает жизненный путь человека. Это основное и самое страшное последствие болезни.
Осложнения со стороны опорно-двигательного аппарата:
- Остеопороз — уменьшение плотности костной ткани,
- Патология суставов — снижение их подвижности из-за сильной мышечной слабости,
- Сколиоз, кифоз, лордоз — различные формы искривления позвоночника.
Поражение органов пищеварения:
- Запоры — результат гиподинамии,
- Потеря веса из-за разрушения мышц,
- Нарушение процесса жевания и глотания требует питания больного через зонд.
Дыхательные расстройства:
- Поверхностное дыхание,
- Слабый кашлевой рефлекс,
- Частые ОРВИ,
- Недостаток кислорода в крови – головные боли по утрам, пробуждения по ночам, слабость, раздражительность, насыщенные сновидения.
У лиц с миопатией Дюшена развивается кардиомиопатия — слабость миокарда, проявляющаяся повышенной утомляемостью, одышкой, отеками ног, перебоями в работе сердца.
Своевременная диагностика и эффективная терапия могут отсрочить развитие осложнений или совсем предотвратить их появление.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики. Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц. Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.
Лечебный процесс
Болезнь Дюшенна, как и любая другая наследственная патология, неизлечима. Врачи проводят симптоматическую терапию, устраняющую неприятные проявления, предупреждающую развитие осложнений и продлевающую жизнь больных.
Комплексное поддерживающее лечение болезни:
- Медикаментозная терапия замедляет, но не останавливает прогрессирование миопатии. Больным назначают глюкокортикостероидные препараты «Преднизолон», «Бетаметазон», сердечные средства «Ри, «Периндоприл», метаболические препараты «Милдронат», «Рибоксин», «Циннаризин», поливитамины «Нейромультивит», «Доппельгерц».
- Физиотерапия заключается в проведении пассивного растяжения пораженных мышц, ЛФК, массажа, электрофореза, гидротерапии, индуктотермии, лазеротерапии, ультрафонофореза.
- Диетотерапия – полноценное питание с достаточным количеством растительных жиров и животных белков. Больным рекомендуют исключить из рациона крепкий чай и кофе, спиртное, пряности, сдобу, шоколад, фаст-фуд. Полезными являются блюда из овощей и фруктов, молочнокислые продукты, крупяные изделия, яйца.
- Чтобы поддержать функционирование органов дыхания на оптимальном уровне, больным необходима искусственная вентиляция легких, насыщающая кровь кислородом. Для этого в домашних условиях используют различные портативные аппараты, а в стационаре тяжелобольным пациентам интубируют трахею или выполняют трахеостомию.
- Ортопедическая помощь необходима всем больным с миопатией Дюшенна. Причем, чем старше становится ребенок, тем ярче выражены признаки патологии, а значит и потребность в ортопедических приспособлениях гораздо больше. Вертикализаторы, инвалидные кресла, специальные шины и корсеты существенно облегчают жизнь больным лицам.
- В отдельных случаях пациентам требуется психотерапевтическая поддержка. Это связано с развитием у детей депрессии и появлением суицидальных наклонностей. Не каждый ребенок в подростковом возрасте может смириться со своей беспомощностью и жизнью в инвалидном кресле.
- Применение стволовых клеток и генная терапия — развивающиеся направления в лечении наследственных заболеваний. Путем введения в организм здоровых генов или замены обычных клеток стволовыми можно улучшить работу организма на клеточном уровне и нормализовать состояние больного. С помощью стволовых клеток восстанавливают пораженные мышечные волокна. Больным подсаживают миогенные клетки, вырабатывающие нормальный дистрофин в достаточном количестве. Любая научная разработка, доказанная опытным путем, дает надежду больным на выздоровление.
Этиология
До конца этиология болезни Дюшенна-Арана заболевания неизвестна. Гипотеза об этнологическом значении дефицита витамина Е не доказана. Наследственные и семейные случаи встречаются только в качестве редкой казуистики. Все же какое-то врожденное предрасположение в виде недостаточной резистентности клеток передних рогов легко допустить, поскольку многие больные обнаруживают черты невропатической конституции. Среди экзогенных моментов, которые могут играть роль в развитии заболевания, указывают на травму центральной нервной системы, различные острые общие инфекции, хронические инфекции, интоксикации. Ставится вопрос и о возможной роли специфического вируса при этой болезни. Подавляющее большинство исследователей рассматривает хронический «полиомиелит» взрослых, лежащий в основе болезни Дюшенна-Арана, как чисто дегенеративный, а не инфекционно-воспалительный процесс. Надо особо подчеркнуть, что никакого отношения к острому полиомиелиту (болезни Гейне-Медина) он не имеет. Все же полностью отрицать значение инфекции при этой болезни нельзя.
Поражение скелетной мускулатуры
Дети появляются на свет без серьезных проблем со здоровьем. Однако уже через несколько месяцев их двигательное развитие начинает отставать. Такие малыши менее активные. Врачи и родители еще не замечают явных отклонений, списывая все на особенности темперамента.
Начальные симптомы заболевания возникают после первых шагов. Дети с дистрофией Дюшенна постоянно падают и передвигаются на носочках. Если большинство сверстников уже уверенно стоит на ногах, они упорно продолжают испытывать трудности с передвижением.
Следующим этапом проявления недуга является период, когда малыши обретают способность к разговору. Они начинают жаловаться родителям на слабость и быструю утомляемость. Прыжки на детской площадке, бег, лазанье по турникам — все эти виды активности не приносят им удовольствия.
Какие еще имеет мышечная дистрофия Дюшенна симптомы? Нарушение Говерса считается своеобразным проявлением заболевания. При каждой попытке встать с пола ребенок пользуется руками, чтобы помочь таким образом слабым мышцам ног. С этой целью он опирается конечностями на себя, перебирая ими по всему телу.
Постепенное прогрессирование недуга приводит к тому, что больные дети к 10-12 годам утрачивают навык самостоятельно передвигаться. Большинство из них нуждается в инвалидном кресле. Способность держать тело в вертикальном положении сохраняется только до 16 лет.
Лечение
Эффективного способа лечения болезни Дюшенна-Арана пока не существует. Проводят несколько курсов дезинфицирующей терапии. При наличии оснований для предположения в прошлом люэса — противосифилитическое лечение. Во всех случаях показаны массаж, неутомительная лечебная физкультура, йодионогальванизация позвоночника, диатермия пострадавших конечностей. Витамины B1, В12, Е. Инъекции АТФ (монокальциевой соли аденозинтрифосфорной кислоты); глютаминовая кислота, гемотрансфузии. Прозерин, дибазол, галантамин.
Патологическая анатомия
Передние и задние рога спинного мозга
Патологоанатомическая картина спинного мозга при болезни Дюшенна-Арана представляет собой медленно прогрессирующую атрофию клеток передних рогов, особенно тех, которые дают начало волокнам передних корешков. С течением времени эти клетки гибнут, продукты их распада выводятся из пределов центральной нервной системы, место ганглиозных клеток занимает невроглия. Волокна передних корешков и их продолжения в периферических нервах подвергаются перерождению и замещаются соединительной тканью. Мышечные волокна, связанные с пораженными сегментами спинного мозга, также атрофируются. На гистологических срезах пострадавшие мышечные волокна лежат небольшими группами (что характерно для неврогенных амиотрофий). Никакой воспалительной сосудисто-соединительнотканной реакции нет. Часто в спинном мозгу при болезни Дюшенна-Арана констатируется утолщение стенок сосудов и сужение просвета их.
Основные причины
Мышечная дистрофия является следствием отклонения в генетическом коде ДНК. Мутация возникает в расположенном в Х-хромосоме гене. Один из его участков отвечает за выработку особого белка — дистрофина. Это вещество на микроскопическом уровне составляет базис мышечных волокон и выполняет несколько функций:
- поддержание клеточного скелета;
- обеспечение способности мышечных волокон к сокращению и расслаблению.
При этом заболевании дистрофин отсутствует или плохо синтезируется. Уровень «нормального» белка не превышает отметки в 3 %. Такая мутация приводит к разрушению волокон в мышцах. Они постепенно перерождаются, замещаются жировой и соединительной тканью. Как следствие, человек утрачивает способность к передвижению.
Какой имеет мышечная дистрофия Дюшенна тип наследования? Заболевание передается по рецессивному признаку. В организме человека все гены парные. Чтобы при наследственном недуге появились патологические нарушения, генетический дефект должен возникнуть в одной хромосоме или в аналогичных участках обеих. Во втором случае речь идет о рецессивном типе наследования.
Если генетический дефект диагностируется только в одной хромосоме, но болезнь при этом прогрессирует, говорят о доминантном признаке передачи. Рецессивный тип возможен при одномоментном поражении одинаковых ДНК-структур. Когда вторая хромосома абсолютно «здорова», патология не развивается. Поэтому дистрофия диагностируется только у лиц мужского пола. У них в генетическом наборе присутствует одна Х-хромосома, а вторая (У) — парная.
Что наука говорит о представительницах прекрасного пола? Мышечная дистрофия Дюшенна у девочек диагностируется редко. Для этого в генотипе должны совпасть две патологические Х-хромосомы, что маловероятно. Девочки могут выступать только носителями недуга и передавать его сыновьям.