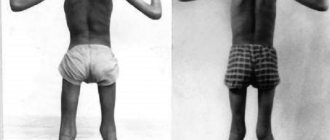
Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Дистрофия или миопатия? Термин «миопатия» является общим названием для всех патологий М. Д. Мышечные дистрофии — это особые формы миопатий. Однако в повседневном языке термин миопатия часто используется для обозначения дегенерации мышц.
Врожденные мышечные дистрофии
Первая группа — это врожденные мышечные дистрофии (нарушение функции и строения мышечных волокон в результате сбоя синтеза белков, которые входят в их состав). Четкой классификации врожденных мышечных дистрофий нет, но на основании гипотез патогенеза данной группы заболевания можно выделить две группы:
- мерозин-негативные заболевания, которые характеризуются дефицитом или отсутствием белка мезонина, входящего в состав поперечно-полосатых мышц;
- врожденные структурные миопатии и мерозин-позитивные, при которых концентрация мезонина находится в норме.
Мерозин-негативная группа заболеваний подразделяется на несколько типов:
- врожденные мышечные дистрофии Фукуямы;
- мышечно-глазо-мозговой синдром;
- синдром Уолкера-Варбурга.
Клиническая картина заболеваний мерозин-негативной группы очень схожа с симптоматикой классических врожденных миопатий, но отличительной особенностью является вовлечение в общую симптоматику различных структур головного мозга, что ведет к дальнейшей умственной отсталости, задержке развития. Заболевания мерозин-позитивной группы намного реже включают в себя поражение центральной нервной системы (приблизительно у 10% больных выявлено поражение мозга) и обычно не влечет за собой торможение интеллекта. Клиническая картина характеризуется деформацией позвоночника и нарушением черт строения лица.
Патологоанатомическая картина заболевания
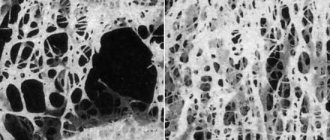
Посмотрим, что происходит внутри мышечных клеток у пациентов, страдающих миодистрофией Дюшенна. Для этого сделаем надрез кожи, расширим расширителем и возьмем небольшой кусочек мышечных волокон.
На фото: биопсия мышечной ткани
На фото: биопсия мышечной ткани
Типичный признак миодистрофии в первую очередь – это разный диаметр мышечных волокон. У здорового человека диаметр мышечных волокон одинаковый.
Характерные признаки мышечной дистрофии – атрофированные и гипертрофированные волокна, множественные внутренние ядра и отек.
Изучая окрашенные срезы скелетной мышцы, я увидел денервацию миофибрилл, значительный разброс в размере миофибрилов и выраженный отек.
Пояснение к первому фото:
- Бледно-фиолетового цвета – это мышечные волокна в разрезе.
- Светлые пятна как внутри волокон, так и снаружи – это отек.
- Темные точки – это ядра, которые отек сместил к периферии.
На втором фото показано нормальное мышечное волокно здорового человека.
Степень тяжести мышечной дистрофии по данным электронной микроскопии ориентируется на следующие показатели:
- при легкой степени разница в размере мышечных волокон выражена умерено, начальные признаки отека (белый цвет).
На фото: биопсия мышечных волокон при легкой (A), средней (B) и тяжелой степени дистрофии (C).
- средняя степень тяжести соответствует перемещению ядер в центр мышечных волокон, расширению межфибриллярного пространства из-за увеличения отека между клетками.
На фото: мышечные волокна при прогрессирующей мышечной дистрофии средней степени тяжести:
а) светло-фиолетового цвета мышечные волокна;
б) светлые пятна внутри мышечных волокон – отек, оттолкнувший ядра из центра клетки к периферии;
в) темные точки – ядра мышечных клеток;
г) стрелкой показана мышечная клетка, которая не может двигаться из-за уменьшения обменных процессов, – темнеет в сторону фиолетового цвета.
- тяжелая степень характеризуется обширными очагами деструкции миофибрилл, их фрагментация и дезорганизация, появление гиалиноподобного вещества и отека между мышечными клетками. Функционально такая ткань обладает слабой силой, быстро наступает утомление и развиваются признаки мышечной усталости. Фото будет представлено немного ниже.
Вот такое состояние мышц было у Эмине до обращения ко мне:
Пояснение к фото “тяжелая степень мышечной дистрофии”:
- Мышечные волокна в разрезе окрашены в синий цвет.
- Красные точки – это ядра мышечных клеток.
- Отек – неокрашенный белый цвет.
Врожденные структурные миопатии
Вторая группа — врожденные структурные миопатии (нарушения целостности цитоскелета мышечных волокон и возникновение патологии в биоптате мышц). Эта группа заболеваний характеризуется нарушением синтеза белков, отвечающих за рост и другие функции формирования мышц в раннем развитии эмбриона.
К врожденным структурным миопатиям относят:
- болезнь центрального стержня;
- немалиновую миопатию;
- центронуклеарную миопатию;
- мегакониальную миопатию;
- миопатию с диспропорцией типов мышечных волокон;
- миопатию со множественными центральными стержнями;
- миотубулярную миопатию;
- миопатию с кристаллическими включениями.
Клинические картины каждого из заболеваний данной группы похожи друг на друга и характеризуются мышечной гипотонией и гипертрофией, пониженной рефлективностью в сухожильях и повышением концентрации в крови креатинфосфокиназы. Наблюдается медленное прогрессирование.
Важно! Регистры (реестры) пациентов с врождённой мышечной дистрофией / миопатией
Регистры пациентов в разных странах вы можете узнать по ссылке: https://www.treat-nmd.eu/resources/patient-registries/list/CMD/.
Международный регистр по врождённым мышечным заболеваниям (CMDIR — Congenital muscle disease international register).
Почему нужно регистрироваться в реестре?
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку генетические нервно-мышечные заболевания являются редкими (орфанными) заболеваниями.
Для этого в разных странах ведутся реестры пациентов — базы данных по генетической и клинической информации о людях, страдающих от генетических нервно-мышечных заболеваний и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных определённым заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Симптомы врожденной миопатии
Врожденная миопатия дебютирует чаще всего в первые месяцы жизни ребенка. Характеризуются данные заболевания наличием синдрома «вялого ребенка»: заметное снижение мышечного тонуса, слабость в мышцах, плохо развивается мускулатура и наблюдается обессиливание во время процесса сосания. С развитием ребенка мышечная слабость более заметно выражена — нехватка сил для того, чтобы стать на ножки или просто поднять свое тело, могут возникнуть трудности при ходьбе или сидении, наблюдается заметное отставание в физическом развитии по сравнению с другими детьми такого же возраста.
Слабость в мышцах может быть выражена сильно или незначительно. Чаще всего симптоматика сохраняется на весь период жизни больного и практически не прогрессирует или слабо развивается. В отдельных случаях, можно наблюдать невозможность самостоятельно передвигаться, поэтому больной вынужден использовать коляску, но навыки самообслуживания, приобретенные им, не утрачиваются.
Врожденные миопатии провоцируют не только слабость мышц конечностей и спины, слабеют и мышцы дыхательной мускулатуры, что является особенно опасным для детей грудного возраста. Если мышечная слабость дыхательных путей выражена в малой степени, то наблюдается развитие дыхательной недостаточности. Это, в свою очередь, провоцирует различные заболевания дыхательных путей (бронхиты, всевозможные виды пневмоний). Иногда дыхательная недостаточность приводит к летальному исходу еще в младенческом возрасте. Бывают случаи, когда с возрастом слабость мышц уменьшается или наоборот прогрессирует.
В отдельных случаях врожденная миопатия проявляется также в виде дисморфичных черт лица (удлиненная форма черепа, высокое небо) или патологиями развития скелета (сколиоз, косолапость, врожденный вывих бедра, кифоз).
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Характеристики наиболее распространенных патологий
Мышечная миопатия Дюшенна. Чаще всего симптомы начинаются примерно в возрасте от 3 до 5 лет. Из-за ослабления мышц ног дети, которые ходили «нормально», часто падают и с трудом встают. Бегать, ходить и прыгать становится для них все труднее. Мышцы, когда они ослабляются, теряют свой объем, за исключением икроножных мышц, которые могут даже увеличиваться путем замены мышечной массы жиром.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Болезнь центрального стержня
Наследуется аутосомно-доминантным способом с неполной пенетрантностью (но встречаются и спорадические случаи наследственности). Данная форма врожденной миопатии характеризуется патологией проксимальных мышц конечностей, но больные способны приобрести некоторые двигательные навыки. В младенческом возрасте наблюдается задержка двигательного развития и гипотония, но диагностировать данное заболевание можно только в более позднем возрасте при изменениях скелета и выраженной мышечной слабости. При этом наблюдаются патологии скелета: деформация стоп, кифосколиоз, дислокация бедер, грудь сапожника.
Чаще всего, больные имеют хрупкую фигуру и невысокий рост. При диагностике заболевания проводят биоптат мышц, который показывает наличие множественных или единичных прерывистых зон, которые лишены ферментов окисления, в некоторых мышечных волокнах. Проведение других лабораторных анализов может показать норму. Пациенты с болезнью центрального стержня склонны к развитию злокачественной гипертермии.
Прогнозы и осложнения
Чаще всего мышечная дистрофия приводит к развитию опасных для жизни осложнений: нарушается работа легких и сердца, сокращается двигательная активность и наступает паралич, искривляется позвоночник, страдают интеллектуальные возможности.
Обнаружение мышечной дистрофии у пациента может стать приговором, но в отдаленной перспективе. Легче всего патологии протекают у взрослых. Если заболевание нашли у ребенка, шанс, что он проживет больше 20 лет, катастрофически мал. Однако поддерживающая терапия способна продлить активную жизнь пациента и минимизировать риск осложнений.
Немалиновая миопатия
Второе название данного заболевания — врожденная непрогрессирующая нитеобразная миопатия. Наследственность в основном передается по аутосомно-доминантному типу, но встречается также рецессивный и спорадический. Возможен летальный исход вследствие дыхательной недостаточности в раннем младенческом возрасте. Наблюдается сильно выраженные патологии скелета. Развитие болезни может происходить в той или иной степени, а может не прогрессировать вовсе. В отдельных случаях больные вынуждены передвигаться с помощью сидячей каталки, в других — страдают от дыхательной недостаточности. При диагностировании проводится гистологическое исследование, которое выявляет в мышцах неподобные или палочкоподобныене малиновые тельца. ЭМГ обычно утверждает диагноз миопатии.
Этиология
Разнородная группа заболеваний. Все они являются наследственными прогрессирующими мышечными дегенеративными процессами, но различаются по своим клиническим и патологическим признакам и типу наследования. Генетический механизм многих из них в настоящее время прояснен (табл. 180-1).
Клинические проявления отдельных заболеваний (см. табл. 180-1)
Дистрофия Дюшенна.
Х-сцепленная мышечная дистрофия, поражающая исключительно мальчиков. Начало заболевания в возрасте до 5 лет; симметричная и неуклонно прогрессирующая слабость в мышцах бедер и плечевого пояса, затрудняющая движения при подъеме, беге, прыжках. К 8-10 годам большинство детей нуждается в ортопедических аппаратах; к 12 годам большинство детей не могут ходить. Больные редко живут более 25 лет.
Сопутствующие нарушения.
Сухожильные и мышечные контрактуры (в том числе ахилловых сухожилий), прогрессирующий кифосколиоз, нарушение функции легких, кардиомиопатия, интеллект снижен. Мышечная слабость сочетается с пальпаторно определяемым увеличением и плотностью некоторых мышц (например, икроножных), что вначале является результатом гипертрофии, а затем замещения мышц жировой и соединительной тканью.
Лабораторные исследования.
Значительное повышение (в 20-100 раз) мышечных ферментов (КФК, альдолаза), миопатическая кривая на ЭМГ; в биоптатах — наличие некротизированных мышечных волокон с регенерацией, фагоцитозом и жировым перерождением мышечной ткани. Диагноз может быть поставлен точно при обнаружении дистрофика в мышечной ткани методом вестерн-блоттинга и (или) иммунохимической метки. Мутации в дистрофин-гене могут быть доказаны примерно у двух третей больных с помощью исследования кДНК. Изменения ЭКГ (увеличенный комплекс RS в отведении V,, глубокий Q в грудных отведениях) свидетельствуют о наличии кардиомиопатии.
Определение носителъства.
Сывороточная КФК повышена у 50% женщин-носителей. Хотя ген и его производное (дистрофии) еще не идентифицированы, в практике можно использовать пробы на кДНК для определения носительства и пре-натальной диагностики.
Осложнения.
Включают дыхательную недостаточность и инфекционные заболевания, аспирацию и острую гастрэктазию. ЗСН и сердечные аритмии также осложняют течение кардиомиопатии. Пассивное растяжение мышц, тенотомия, пш-нирование, физиотерапия, вспомогательные механические устройства и отказ от длительной иммобилизации — все эти меры симптоматической терапии могут быть полезны.
Таблица 180-1 Прогрессирующие мышечные дистрофии
Тип
| Генетический механизм | Клинические признаки | Вовлечение других систем органов | |
| Дюшенна | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в возрасте до 5 лет; прогрессирующая слабость мышц тазового и плечевого пояса; неспособность ходить после 12 лет; кифосколиоз; дыхательная недостаточность в возрасте 20-30 лет | Кардиомиопатия; снижение интеллекта |
| Беккера | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в раннем или позднем возрасте; медленно прогрессирующая слабость мышц тазового и плечевого пояса; сохранение способности ходить после 15 лет; дыхательная недостаточность после 40 лет | Кардиомиопатия |
| Миотоническая | Аутосомно- доминантный; расширение нестабильного участка ДНК хромосомы 19ql3,3 | Начало в любом возрасте; медленно прогрессирующая слабость мышц век, лица, шеи, дистальных мышц конечностей; миотония | Нарушение сердечной проводимости; психические нарушения; катаракты, лобная алопеция; атрофия гонад |
Таблица 180-1
Продолжение
| Тип | Генетический механизм | Клинические признаки | Вовлечение других систем органов |
| Плече-лопаточно-лицевая | Аутосомно-доминантный; часто мутации хромосомы 4q35 | Начало в возрасте до 20 лет; медленно прогрессирующая мышечная слабость лицевой области, плечевого пояса, тыльного сгибания стопы | Гипертензия; глухота |
| Плечевого и тазового пояса (возможны несколько заболеваний) | Аутосомно-рецессивный или доминантный | Начало с раннего детства до среднего возраста; медленно прогрессирующая слабость мышц плечевого и тазового пояса | Кардиомиопатия |
| Глазо-глоточная | Аутосомно-доминантный (Французская Канада или Испания) | Начало в 50-60 лет; медленно прогрессирующая слабость мышц: наружных глазных, век, лица и глотки; крикофарингеальная ахалазия. | Церебральные, глазные |
| Врожденная (включает несколько заболеваний, в том числе типы Фукуяма ицеребро-окулярная дисплазия) | Аутосомно-рецессивный | Начало при рождении; гипотония, контрактуры, задержка развития; в одних случаях — ранняя дыхательная недостаточность, в других — более благоприятное течение болезни |
Источник: Mendell JR, Griggs RC, НРШ-13, с. 2384.
Лечение.
Пока не разработано. Преднизон, 0,75 мг/кг/сут может замедлить про-грессирование заболевания на период до 3 лет.
Дистрофия Беккера (доброкачественная псевдогипертрофическая).
Менее тяжелая и реже встречающаяся, чем дистрофия Дюшенна, с более медленным течением и более поздним началом (5-15 лет), но со сходными клиническими и лабораторными признаками. Это заболевание также является результатом дефекта в ди-строфин-гене.
Миотоническая дистрофия.
Аутосомно-доминантное заболевание, при котором мышечная слабость проявляется в возрасте 20-30 лет, с первоначальным вовлечением мышц лица, шеи и дистальных мышц конечностей. Это приводит к возникновению типичной внешности, характеризующейся птозом, гипотрофией височных областей, отвисшей нижней губой и опущенной нижней челюстью. Миотония проявляется в характерной неспособности больного быстро расслабить мышцы после сильного напряжения (например, после плотного сжатия кисти в кулак) и длительном сокращении мышц после постукивания (например, по языку или возвышению большого пальца кисти).
Сопутствующие нарушения:
алопеция лобной части, задняя субкапсулярная катаракта, атрофия гонад, дыхательные и сердечные нарушения, эндокринная патология, интеллектуальные расстройства и гиперсомния.
Лабораторные исследования.
Уровень КФК нормален или слегка повышен, характерные признаки миотонии и миопатии на ЭМГ, типичные признаки повреждения волокон в мышечных биоптатах. Осложнения со стороны сердца, включая полную сердечную блокаду, представляют серьезную угрозу жизни больного. Следует тщательно контролировать дыхательную функцию, так как хроническая гипоксия может вести к развитию легочного сердца.
Ранняя диагностика.
У больных имеется нестабильный участок ДНК с повышенным количеством CTG-триплетных повторов в хромосомном локусе 19ql3.3. Молекулярно-генетические исследования способствуют раннему выявлению и пре-натальной диагностике.
Лечение.
Фенитоин, прокаинамид, хинин применяются в лечении миотонии, но требуется осторожность у больных с заболеваниями сердца (опасность ухудшения сердечной проводимости). Имплантация водителя сердечного ритма необходима больным с синкопе или сердечной блокадой. Применение ортопедических аппаратов может укрепить «висячие» стопы, стабилизировать голеностопные суставы, уменьшить частоту падений.
Плечелопаточно-лицевая дистрофия.
Обычно медленно прогрессирующее, умеренной тяжести заболевание, начало которого приходится на возраст 30-40 лет. Развивается мышечная слабость лица, плечевого пояса и проксимальных мышц рук, что сопровождается атрофией двуглавых и трехглавых мышц, появлением «крыловидных лопаток» и скошенных плеч. Слабость мимической мускулатуры выражается в неспособности свистеть и потере выразительности лица. Свисание стоп и слабость ног может вызывать падения больного и прогрессирующее затруднение движений.
Лабораторные исследования.
Уровень КФК нормален или слегка повышен, смешанные признаки миопатии-невропатии на ЭМГ и в биоптатах мышц. У больных определяются мутации в хромосоме 4q35. Специфическое для этого локуса генетическое исследование проводят для выявления носительства и в пренатальной диагностике. Ортопедические средства и другие стабилизационные меры могут быть полезны для отдельных больных.
Реже встречающиеся дистрофии
Лопаточно-перонеальная дистрофия.
Клиническая картина такая же, как при плечелопаточно-лицевой дистрофии, но нет мышечной слабости лица, возможны явления кардиомиопатии. В большинстве случаев заболевание начинается в среднем возрасте и наследуется по аутосомно-доминантному типу, но может встречаться и форма болезни с ранним началом, другим механизмом генетической передачи (связанный с Х-хромосомой, рецессивный), с клиническими проявлениями суставных контрактур и кардиомиопатией (тип Эмери—Дрейфуса).
Глазоглоточная дистрофия
(прогрессирующая наружная офтальмоплегия). Начало заболевания в возрасте 50-60 лет с явлениями птоза, ограничения экстраокулярных движений, лицевой и крикофарингеальной мышечной слабости. Крикофа-рингеальная мышечная слабость ведет к ахалазии, дисфагии и аспирации. Так как нарушения движений глаз носят хронический характер, они редко ведут к дипло-пии. Большинство больных испанского или франко-канадского происхождения.
Дистрофия мышц поясов конечностей.
Возможно, представляет собой группу заболеваний, ключевым симптомом которых является слабость проксимальных мышц верхних и нижних конечностей. В зависимости от конкретного подтипа наблюдаются различия в возрасте больных, на который приходится начало болезни, скорости прогрессирования, тяжести клинических проявлений и в сопутствующих осложнениях (сердечные, дыхательные). Лабораторные показатели включают: повышенный уровень КФК и признаки миопатии на ЭМГ и в биоптатах мышц.
Таблица 180-2 Токсические миопатии
Очаговые (фокальные) миопатии: пентазоцин, меперидин, героин. Генерализованные миопатии: А. Воспалительные: циметидин, D-пеницилламин, прокаинамид Б. Мышечная слабость и миалгии: зидовудин, хлорохин, клофибрат, колхицин, глюкокортикоиды, эметин, аминокапроновая кислота, лабеталол, пергексилен, пропанолол, винкристин, ниацин, циклоспорин В. Рабдомиолиз и миоглобинурия: алкоголь, героин, амфетамин, клофибрат, ловастатин, гемфиброзил, аминокапроновая кислота, фенциклидин, барбитураты, кокаин Г. Злокачественная гипертермия: галотан, этилен, диэтилэфир, метоксилфлуран, этилхлорид, трихлорэтилен, галламин, суксинилхолин, лидокаин, мепивакаин Источник: Mendell JR, Griggs RC, HPIM-13, p. 2392.
Дистальная дистрофия.
Группа редко встречающихся заболеваний, среди которых выделяют несколько вариантов с началом в различном возрасте и разным типом наследования. Обычно проявляются мышечной слабостью кистей и стоп с медленным распространением на проксимальные мышечные группы. Повышен уровень КФК; признаки миопатии на ЭМГ и в биоптатах мышц.
Метаболические миопатии
Возникают в результате нарушения утилизации мышцами глюкозы и жирных кислот как источников энергии. У больных определяется либо острый синдром миалгии, миолиза и миоглобинурии, либо хронически прогрессирующая мышечная слабость. Для точной диагностики необходимо провести биохимическое и ферментативное исследование биоптатов мышц. Тем не менее мышечные ферменты, ЭМГ и биоптаты мышц, как правило, выявляют патологические изменения, и на их основании можно заподозрить конкретное заболевание.
При детских и младенческих формах гликогеновой болезни часто определяются сопутствующие расстройства с нарушением функции сердца, печени и эндокринной системы, симптомы которых могут затруднить диагностику мышечных нарушений. Формы, встречающиеся у детей и взрослых, могут имитировать мышечную дистрофию или полимиозит. При некоторых типах заболевания его проявлением может быть один из приступов мышечных судорог или мышечная усталость, спровоцированная нагрузкой. Лактат-тест ишемии предплечья имеет диагностическую ценность, так как отсутствует повышение уровня молочной кислоты в сыворотке после нагрузки (характерного для нормы). Нарушение метаболизма жирных кислот проявляется клиническими признаками, сходными с описанными выше. У некоторых больных определяются мышечные судороги, спровоцированные нагрузкой, мио-лиз и миоглобинурия; у других клиническая картина напоминает полимиозит или мышечную дистрофию. Иногда эффективны специальная диета (включающая жирные кислоты с цепочками средней длины, обогащенными триглицеридами), добавление карнитина внутрь или глюкокортикоиды.
Прочие миопатии
Миопатии могут быть связаны с эндокринными нарушениями, особенно с гипо-или гиперфункцией щитовидной железы, околощитовидных желез и надпочечников. Лекарственные препараты (особенно глюкокортикоиды) и определенные токсины (в том числе алкоголь) — частые причины миопатии (табл. 180-2); в большинстве случаев мышечная слабость бывает симметричной и поражает проксимальные мышцы поясов конечностей. Частые симптомы: мышечная слабость, миалгия, судороги. Диагностика часто зависит от трактовки признаков миопатии при лечении основного заболевания или устранении этиологического агента, так как лабораторные исследования мышечных ферментов, ЭМГ, и даже мышечных биоптатов, не дают специфических данных для диагноза.
Диагностика врожденной миопатии
Диагностика при врожденных миопатиях — сложный процесс, поскольку врачу необходимо дифференцировать и определить конкретный вид миопатии для назначения адекватного лечения и постановки правильного диагноза. Врач-невропатолог выявляет неврологические симптомы, проводит электрофизиологическое и биохимическое исследование, чтобы установить гетерозиготное носительство миопатического гена. Электромиографическое исследование с помощью накожных электродов показывает зачастую снижение вольтажа кривой ЭМГ. При биохимическом анализе крови в сыворотке наблюдается повышенная концентрация альдолазы и креатинкиназы.
Диагностика
- ДНК-диагностика.
- Исследование сыворотки крови (повышение КФК в 5–20 раз, ЛДГ).
- ЭМГ (первично-мышечный характер изменений).
- Биопсия скелетных мышц (признаки первичной мышечной дистрофии и денервации).
- ЭКГ/ЭхоКГ (нарушение атриовентрикулярной и/или внутрижелудочковой проводимости, гипертрофия миокарда, дилатация желудочков, застойная сердечная недостаточность, кардиомиопатия).
Дифференциальный диагноз:
- Прогрессирующая мышечная дистрофия Дюшенна.
- Мышечная дистрофия Эрба.
- Спинальная амиотрофия Кугельберга-Веландер.
- Полимиозит, дерматомиозит.
- Метаболическая миопатия.
- Наследственные полиневропатии.
Лечение врожденной миопатии
Лечение врожденный миопатий малоэффективное. Четкого лечения на данный момент нет. О том, можно ли лечить врожденную миопатию, ученые спорят до сих пор. Медицинские институты разных стран проводят исследования на генном уровне — с использованием стволовых клеток. Существует симптоматическое лечение, которое заключается в воздействии на обменные процессы в организме пациента, в частности, синтез белков, попытку нормализации функций вегетативной нервной системы. Чаще всего медикаментозное лечение включает в себя принятие анаболических гормонов (неробол, цераксон, ретаболил, сомазин), АТФ. Обязательно проводится витаминотерапия. Назначаются также антихолинэстеразные препараты.
Обязательным звеном процесса лечения врожденных миопатий является лечебная физкультура. Это могут быть занятия в воде или комплекс упражнений. ЛФК контролируется тренером или неврологом. В отдельных случаях действенной оказывается ортопедическая коррекция (например, ношение ортопедической обуви, корсетов или использование ортопедических матрасов, подушек, кресел).
Состояние и клиническую картину заболевания контролирует невропатолог, терапевт, педиатр, кардиолог и ортопед-травматолог.
Как лечить дистрофию мышц?
Наследственное заболевание достаточно сложно поддается лечению и корректировке. Но современная медицина предлагает в качестве лечения мероприятия, способствующие минимизации влияния болезни на организм человека.
Прогрессия болезни может дать осложнения с позвоночником, привести к его деформации, также это развитие проблем с дыханием и частые проявления пневмонии. Мышечная недостаточность отражается на главном органе человека — сердце, ведь это тоже мышца.
Терапия может улучшить качество жизни пациентов за счет укрепления сердца, костного корсета и легких. Мышечная дистрофия взрослых и детей лечится следующим образом:
- Употребление пациентом для накопления энергии и снижения симптомов болезни кортикостероидов.
- Назначение физических нагрузок – лечебная физическая культура, плаванье, так как отсутствие активности мышц приводит к более быстрому прогрессу заболевания.
- Назначается физиотерапия, направленность которой — это поддержание тонуса в мышцах, улучшение функционирования суставов. Также пациент применяет специальные дыхательные гимнастики.
- Пациенту назначается использование различных ортопедических приспособлений.
Данные мероприятия могут быть слабо эффективными и не помогать в сопротивлении развития болезни. При нарушении дыхательной системы пациенту поддерживают дыхание при помощи респираторных механизмов.
Медицина сейчас развивается, и появляются новые методы лечения, которые борются с прогрессивным течением болезни. Такими нововведениями является лечение стволовыми клетками, замена или восстановление дистрофина (белка, который отвечает за поддержание функций скелетных и сердечных мышц), создание противовоспалительных препаратов с минимальным возникновением побочных эффектов и средств, направленных на улучшение мышечной функциональности.
Последней инновацией, которая предоставляет абсолютное излечение от болезни, является редактирование генов. Данный метод находится в разработке, и неизвестно, когда он перейдет на стадию практического применения.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики. Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц. Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.
Причина заболевания
В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Заболевание наследуется по рецессивному типу, сцепленному с Х хромосомой. Что это означает? Поскольку все гены человека парные, то есть дублируют друг друга, то для того, чтобы появились патологические изменения в организме при наследственном заболевании, необходимо, чтобы генетический дефект возник в одной хромосоме или аналогичных участках обеих хромосом. Если заболевание возникает только при мутациях в обеих хромосомах, то такой тип наследования называют рецессивным. Когда же генетическая аномалия выявляется только в одной хромосоме, но болезнь все равно развивается, такой тип наследования называют доминантным. Рецессивный тип возможен только при одновременном поражении идентичных хромосом. Если вторая хромосома будет «здорова», то заболевание не возникнет. Именно поэтому мышечная дистрофия Дюшенна является уделом лиц мужского пола, потому что они имеют в генетическом наборе одну Х хромосому, а вторую (парную) – У. Если мальчику попадается «поломанная» Х хромосома, то у него обязательно возникает болезнь, потому что здоровой хромосомы у него просто нет. Для того, чтобы мышечная дистрофия Дюшенна возникла у девочки, необходимо совпадение в ее генотипе двух патологических Х хромосом, что практически маловероятно (в таком случае папа девочки должен быть болен, а у мамы в генетическом наборе должна содержаться дефектная Х хромосома). Девочки выступают лишь носителями заболевания и передают его своим сыновьям. Конечно, часть случаев заболевания не является результатом передачи по наследству, а возникает спорадически. Это означает появление мутации в генетическом наборе ребенка спонтанно. Вновь появившаяся мутация может быть передана по наследству (при условии сохранения способности к размножению).