Muscular dystrophy (MD) is a group of diseases characterized by progressive weakness and muscle degeneration. The muscles gradually atrophy - they lose their volume and, therefore, strength.
These are diseases of genetic origin that can occur at any age: from birth, in childhood or in adulthood. There are more than 30 forms of the disease, which vary in age at onset of symptoms, the type of muscles affected and severity. Most types of dystrophies gradually become more complicated and have irreversible consequences. Currently, there is still no cure for MD. The most famous and common type of disease is Duchenne myopathy.
During the development of MD, the primary muscles that are affected are those that facilitate voluntary movement, including the hips, legs, arms, and forearms. In some cases, the respiratory muscles and heart may be affected. People with muscular dystrophy gradually lose their mobility when walking. Other symptoms may be related to muscle weakness, including heart, gastrointestinal, and eye problems.
Dystrophy or myopathy? The term “myopathy” is a common name for all MD pathologies. Muscular dystrophies are special forms of myopathies. However, in everyday language, the term myopathy is often used to refer to muscle degeneration.
Congenital muscular dystrophies
The first group is congenital muscular dystrophy (impaired function and structure of muscle fibers as a result of a failure in the synthesis of proteins that make up their composition). There is no clear classification of congenital muscular dystrophies, but based on the hypotheses of the pathogenesis of this group of diseases, two groups can be distinguished:
- merosin-negative diseases, which are characterized by a deficiency or absence of the mezzanine protein, which is part of striated muscles;
- congenital structural myopathies and merosin-positive, in which the mezzanine concentration is normal.
The merosin-negative group of diseases is divided into several types:
- Fukuyama congenital muscular dystrophies;
- muscular-eye-brain syndrome;
- Walker-Warburg syndrome.
The clinical picture of diseases of the merosin-negative group is very similar to the symptoms of classical congenital myopathies, but a distinctive feature is the involvement of various brain structures in the general symptoms, which leads to further mental retardation and developmental delay. Diseases of the merosin-positive group much less often involve damage to the central nervous system (approximately 10% of patients have brain damage) and usually do not entail inhibition of intelligence. The clinical picture is characterized by spinal deformation and disturbance of facial features.
Pathological picture of the disease
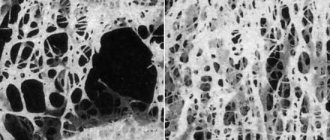
Let's see what happens inside muscle cells in patients suffering from Duchenne muscular dystrophy. To do this, we will make an incision in the skin, expand it with an expander and take a small piece of muscle fiber.
In the photo: muscle tissue biopsy
In the photo: muscle tissue biopsy
A typical sign of muscular dystrophy is primarily a different diameter of muscle fibers. In a healthy person, the diameter of the muscle fibers is the same.
Characteristic signs of muscular dystrophy are atrophied and hypertrophied fibers, multiple internal nuclei and edema.
Examining stained sections of skeletal muscle, I observed denervation of myofibers, significant variation in myofibril size, and significant edema.
Explanation for the first photo:
- The pale purple color is muscle fibers in cross-section.
- Light spots both inside and outside the fibers are swelling.
- Dark dots are nuclei that the edema has shifted to the periphery.
The second photo shows normal muscle fiber from a healthy person.
The severity of muscular dystrophy according to electron microscopy is based on the following indicators:
- with a mild degree, the difference in the size of muscle fibers is moderate, the initial signs of edema (white color).
In the photo : muscle fiber biopsy for mild (A), moderate (B) and severe dystrophy (C).
- moderate severity corresponds to the movement of nuclei to the center of muscle fibers, expansion of the interfibrillar space due to increased edema between the cells.
In the photo: muscle fibers with progressive muscular dystrophy of moderate severity:
a) light purple muscle fibers;
b) light spots inside the muscle fibers - swelling, which has pushed the nuclei from the center of the cell to the periphery;
c) dark dots – muscle cell nuclei;
d) the arrow shows a muscle cell that cannot move due to a decrease in metabolic processes - it darkens towards purple.
- severe degree is characterized by extensive foci of destruction of myofibrils, their fragmentation and disorganization, the appearance of a hyaline-like substance and edema between muscle cells. Functionally, such tissue has weak strength, fatigue sets in quickly and signs of muscle fatigue develop. The photo will be presented a little lower.
This is the state of Emine’s muscles before contacting me:
Explanation for the photo “severe muscular dystrophy”:
- Muscle fibers in section are colored blue.
- The red dots are the nuclei of muscle cells.
- Edema is uncolored white.
Congenital structural myopathies
The second group is congenital structural myopathies (violations of the integrity of the cytoskeleton of muscle fibers and the occurrence of pathology in muscle biopsy). This group of diseases is characterized by a violation of the synthesis of proteins responsible for growth and other functions of muscle formation in the early development of the embryo.
Congenital structural myopathies include:
- central core disease;
- nemaline myopathy;
- centronuclear myopathy;
- megaconial myopathy;
- myopathy with disproportion of muscle fiber types;
- myopathy with multiple central cores;
- myotubular myopathy;
- myopathy with crystalline inclusions.
The clinical pictures of each of the diseases in this group are similar to each other and are characterized by muscle hypotonia and hypertrophy, decreased reflectivity in the tendons and increased concentrations of creatine phosphokinase in the blood. Slow progression is observed.
Important! Registers of patients with congenital muscular dystrophy/myopathy
You can find patient registries in different countries at the link: https://www.treat-nmd.eu/resources/patient-registries/list/CMD/.
International register of congenital muscle diseases (CMDIR - Congenital muscle disease international register).
Why do you need to register in the registry?
As new drugs are developed, there is a need to test them in clinical settings, and sometimes it takes years to find the right number of patients for studies because genetic neuromuscular diseases are rare (orphan) diseases.
To achieve this, different countries maintain patient registries - databases of genetic and clinical information on people suffering from genetic neuromuscular diseases and who want to speed up the research process. The register allows specialists to obtain information about the condition and number of patients with a certain disease. This information contributes to the development and improvement of patient care standards. It is used to find participants for clinical trials and also to help specialists gain more information about a disease.
Symptoms of congenital myopathy
Congenital myopathy most often debuts in the first months of a child’s life. These diseases are characterized by the presence of the “flaccid baby” syndrome: a noticeable decrease in muscle tone, muscle weakness, poor muscle development and weakness during the sucking process. As the child develops, muscle weakness is more noticeable - lack of strength to stand on his feet or simply lift his body, difficulties may arise when walking or sitting, and there is a noticeable lag in physical development compared to other children of the same age.
Muscle weakness can be severe or mild. Most often, symptoms persist for the entire period of the patient’s life and practically do not progress or develop weakly. In some cases, it is possible to observe the inability to move independently, so the patient is forced to use a wheelchair, but the self-care skills acquired by him are not lost.
Congenital myopathies provoke not only weakness of the muscles of the limbs and back, but also the muscles of the respiratory muscles weaken, which is especially dangerous for infants. If muscle weakness of the respiratory tract is expressed to a small extent, then the development of respiratory failure is observed. This, in turn, provokes various respiratory diseases (bronchitis, all kinds of pneumonia). Sometimes respiratory failure leads to death in infancy. There are cases when muscle weakness decreases with age or, on the contrary, progresses.
In some cases, congenital myopathy also manifests itself in the form of dysmorphic facial features (elongated skull shape, high palate) or pathologies of skeletal development (scoliosis, clubfoot, congenital hip dislocation, kyphosis).
Symptoms of the disease
MD is characterized by muscle weakness that tends to gradually worsen; symptoms vary depending on the type of pathology. Depending on the case, other symptoms such as cardiac and respiratory disorders, eye abnormalities (malformations, cataracts), intellectual deficit, hormonal disorders, etc. may be present.
Characteristics of the most common pathologies
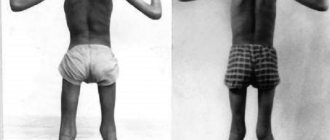
Duchenne muscular myopathy . Symptoms most often begin around the ages of 3 to 5 years. Due to weakened leg muscles, children who walked “normally” often fall and have difficulty standing up. Running, walking and jumping are becoming increasingly difficult for them. Muscles, when weakened, lose their volume, with the exception of the calf muscles, which can even become larger by replacing muscle mass with fat.
Children often complain of cramps and muscle pain. The disease develops quite quickly as soon as the first symptoms appear. Wheelchair use is usually required around the age of 12 years. This type of disorder leads to scoliosis and joint deformities. In addition, some children have mental retardation. By the end of adolescence, cardiac complications (heart failure) and respiratory problems requiring artificial air supply are common. Average life expectancy (from 20 to 30 years on average).
Becker's myopathy .
Symptoms are comparable to those of Duchenne MD, however they are less pronounced and the progression of the disease is slower. Symptoms begin between 5 and 15 years of age, sometimes later, and are characterized by a progressive loss of muscle strength in the limbs and surrounding torso. In more than half of cases, walking remains possible until age 40.
Central core disease
It is inherited in an autosomal dominant manner with incomplete penetrance (but sporadic cases of heredity also occur). This form of congenital myopathy is characterized by pathology of the proximal muscles of the limbs, but patients are able to acquire some motor skills. In infancy, delayed motor development and hypotonia are observed, but this disease can only be diagnosed at a later age with skeletal changes and severe muscle weakness. In this case, skeletal pathologies are observed: foot deformation, kyphoscoliosis, hip dislocation, shoemaker's chest.
Most often, patients have a fragile figure and short stature. When diagnosing the disease, a muscle biopsy is performed, which shows the presence of multiple or single discontinuous zones that are devoid of oxidative enzymes in some muscle fibers. Other laboratory tests may show normal results. Patients with central core disease are prone to developing malignant hyperthermia.
Prognosis and complications
Most often, muscular dystrophy leads to the development of life-threatening complications: the functioning of the lungs and heart is disrupted, motor activity is reduced and paralysis occurs, the spine is bent, and intellectual capabilities are affected.
The discovery of muscular dystrophy in a patient can be a death sentence, but in the long term. Pathologies occur most easily in adults. If the disease is found in a child, the chance that he will live more than 20 years is catastrophically small. However, maintenance therapy can prolong the patient’s active life and minimize the risk of complications.
Nemaline myopathy
The second name for this disease is congenital non-progressive filamentous myopathy. Heredity is mainly transmitted in an autosomal dominant manner, but recessive and sporadic ones are also found. Possible death due to respiratory failure in early infancy. Severe skeletal pathologies are observed. The development of the disease may occur to varying degrees, or may not progress at all. In some cases, patients are forced to move using a sitting gurney, in others they suffer from respiratory failure. When diagnosing, a histological examination is carried out, which reveals unusual or rod-like, non-crimson bodies in the muscles. EMG usually confirms the diagnosis of myopathy.
Chapter 180Myopathies and muscular dystrophiesEtiology
A heterogeneous group of diseases. All of them are hereditary progressive muscle degenerative processes, but differ in their clinical and pathological features and mode of inheritance. The genetic mechanism of many of them has now been clarified (Table 180-1).
Clinical manifestations of individual diseases (see Table 180-1)
Duchenne dystrophy.
X-linked muscular dystrophy, which exclusively affects boys. Onset of the disease before the age of 5 years; symmetrical and steadily progressive weakness in the muscles of the hips and shoulder girdle, making it difficult to move when lifting, running, and jumping. By the age of 8-10, most children require orthopedic devices; By age 12, most children cannot walk. Patients rarely live more than 25 years.
Associated disorders.
Tendon and muscle contractures (including Achilles tendons), progressive kyphoscoliosis, impaired pulmonary function, cardiomyopathy, reduced intelligence. Muscle weakness is combined with a palpable increase and density of some muscles (for example, gastrocnemius), which is initially the result of hypertrophy and then replacement of muscles with fat and connective tissue.
Laboratory research.
Significant increase (20-100 times) of muscle enzymes (CPK, aldolase), myopathic EMG curve; in biopsy samples - the presence of necrotic muscle fibers with regeneration, phagocytosis and fatty degeneration of muscle tissue. The diagnosis can be made accurately when dystrophy is detected in muscle tissue using Western blotting and (or) immunochemical labeling. Mutations in the dystrophin gene can be proven in about two thirds of patients using cDNA testing. ECG changes (enlarged RS complex in lead V, deep Q in the chest leads) indicate the presence of cardiomyopathy.
Determination of carrier status.
Serum CPK is elevated in 50% of female carriers. Although the gene and its derivative (dystrophies) have not yet been identified, cDNA tests can be used in practice to determine carrier status and prenatal diagnosis.
Complications.
Includes respiratory failure and infectious diseases, aspiration and acute gastrectasis. CHF and cardiac arrhythmias also complicate the course of cardiomyopathy. Passive muscle stretching, tenotomy, pinning, physical therapy, mechanical assistive devices, and avoidance of prolonged immobilization are all symptomatic measures that may be helpful.
Table 180-1 Progressive muscular dystrophies
Type
| Genetic mechanism | Clinical signs | Involvement of other organ systems | |
| Duchenne | X-chromosomal recessive mutation of the dystrophin gene | Onset before age 5 years; progressive weakness of the muscles of the pelvic and shoulder girdle; inability to walk after age 12; kyphoscoliosis; respiratory failure at the age of 20-30 years | Cardiomyopathy; decreased intelligence |
| Becker | X-chromosomal recessive mutation of the dystrophin gene | Onset at early or late age; slowly progressive weakness of the muscles of the pelvic and shoulder girdle; maintaining the ability to walk after 15 years; respiratory failure after 40 years | Cardiomyopathy |
| Myotonic | Autosomal dominant; expansion of the unstable DNA region of chromosome 19ql3,3 | Onset at any age; slowly progressive weakness of the muscles of the eyelids, face, neck, distal muscles of the limbs; myotonia | Cardiac conduction disturbances; mental disorders; cataracts, frontal alopecia; gonadal atrophy |
Table 180-1
Continued
| Type | Genetic mechanism | Clinical signs | Involvement of other organ systems |
| Shoulder-scapulofacial | Autosomal dominant; often mutations of chromosome 4q35 | Onset before age 20; slowly progressive muscle weakness of the face, shoulder girdle, and dorsiflexion of the foot | Hypertension; deafness |
| Shoulder and pelvic girdle (several diseases are possible) | Autosomal recessive or dominant | Beginning in early childhood to middle age; slowly progressive weakness of the shoulder and pelvic girdle muscles | Cardiomyopathy |
| Oculopharyngeal | Autosomal dominant (French Canada or Spain) | Onset at 50-60 years old; slowly progressive muscle weakness: external eyelids, eyelids, face and pharynx; cricopharyngeal achalasia. | Cerebral, ocular |
| Congenital (includes several diseases, including Fukuyama types and cerebro-ocular dysplasia) | Autosomal recessive | Onset at birth; hypotension, contractures, developmental delay; in some cases - early respiratory failure, in others - a more favorable course of the disease |
Source: Mendell JR, Griggs RC, NRS-13, p. 2384.
Treatment.
Not developed yet. Prednisone, 0.75 mg/kg/day can slow the progression of the disease for up to 3 years.
Becker's dystrophy (benign pseudohypertrophic).
Less severe and less common than Duchenne dystrophy, with a slower course and later onset (5-15 years), but with similar clinical and laboratory features. This disease also results from a defect in the dystrophin gene.
Myotonic dystrophy.
An autosomal dominant disorder in which muscle weakness appears between the ages of 20 and 30, with initial involvement of the muscles of the face, neck and distal muscles of the extremities. This leads to a typical appearance, characterized by ptosis, hypotrophy of the temporal regions, drooping lower lip and drooping lower jaw. Myotonia is manifested in the patient's characteristic inability to quickly relax muscles after strong tension (for example, after tightly clenching the hand into a fist) and prolonged muscle contraction after tapping (for example, on the tongue or the eminence of the thumb).
Associated disorders:
frontal alopecia, posterior subcapsular cataract, gonadal atrophy, respiratory and cardiac disorders, endocrine pathology, intellectual disorders and hypersomnia.
Laboratory research.
CPK levels are normal or slightly elevated, characteristic signs of myotonia and myopathy on EMG, typical signs of fiber damage in muscle biopsies. Cardiac complications, including complete heart block, pose a serious threat to the patient's life. Respiratory function should be carefully monitored, as chronic hypoxia can lead to the development of cor pulmonale.
Early diagnosis.
Patients have an unstable DNA region with an increased number of CTG triplet repeats in the chromosomal locus 19ql3.3. Molecular genetic studies contribute to early detection and prenatal diagnosis.
Treatment.
Phenytoin, procainamide, quinine are used in the treatment of myotonia, but caution is required in patients with heart disease (risk of worsening cardiac conduction). Pacemaker implantation is necessary for patients with syncope or heart block. The use of orthopedic devices can strengthen “drop” feet, stabilize ankle joints, and reduce the frequency of falls.
Humerofacial dystrophy.
Usually a slowly progressive, moderately severe disease, the onset of which occurs at the age of 30-40 years. Muscular weakness of the face, shoulder girdle and proximal muscles of the arms develops, which is accompanied by atrophy of the biceps and triceps muscles, the appearance of “winged shoulder blades” and sloping shoulders. Weakness of the facial muscles is expressed in the inability to whistle and loss of facial expression. Dropping of the feet and weakness of the legs can cause the patient to fall and cause progressive difficulty in movement.
Laboratory research.
CK levels are normal or slightly elevated, mixed signs of myopathy-neuropathy on EMG and muscle biopsy. Patients are diagnosed with mutations in chromosome 4q35. Genetic research specific to this locus is carried out to identify carriage and in prenatal diagnosis. Orthotics and other stabilization measures may be helpful in selected patients.
Less common dystrophies
Scapuloperoneal dystrophy.
The clinical picture is the same as with glenohumeral-facial dystrophy, but there is no facial muscle weakness, and cardiomyopathy is possible. In most cases, the disease begins in middle age and is inherited in an autosomal dominant manner, but a form of the disease with an early onset, a different mechanism of genetic transmission (X-linked, recessive), with clinical manifestations of joint contractures and cardiomyopathy (Emery type) may also occur. —Dreyfus).
Oculopharyngeal dystrophy
(progressive external ophthalmoplegia). The onset of the disease is at the age of 50-60 years with symptoms of ptosis, limitation of extraocular movements, facial and cricopharyngeal muscle weakness. Cricopharyngeal muscle weakness leads to achalasia, dysphagia, and aspiration. Since eye movement disorders are chronic, they rarely lead to diplopia. Most patients are of Spanish or French-Canadian origin.
Dystrophy of the muscles of the limb girdles.
Possibly a group of diseases in which the key symptom is weakness of the proximal muscles of the upper and lower extremities. Depending on the specific subtype, differences are observed in the age of patients at the onset of the disease, the rate of progression, the severity of clinical manifestations and in associated complications (cardiac, respiratory). Laboratory findings include elevated CPK levels and signs of myopathy on EMG and muscle biopsies.
Table 180-2 Toxic myopathies
Focal myopathies: pentazocine, meperidine, heroin. Generalized myopathy: A. Inflammatory: Qimetidine, D-Penicilllamine, Prokainamide B. Muscle weakness and myalgia: Zidovudin, Chlorochin, Clophibrat, Colchicin, Glucocorticoids, Emetin, Aminocaproneic acid, Labetalol, Pergexile, Prophanolol, Vincristine, Niazin, Tsilosorin V. Rabdor. myolis and myoglobinuria: alcohol, heroin, amphetamine, clofibrate, lovastatin, gemfibrozil, aminocaproic acid, phencyclidine, barbiturates, cocaine D. Malignant hyperthermia: halothane, ethylene, diethyl ether, methoxyflurane, ethyl chloride, trichlorethylene, gallamine, suxinylcholine, lidocaine, mepiva Cain Source: Mendell JR, Griggs RC, HPIM-13, p. 2392.
Distal dystrophy.
A group of rare diseases, among which there are several variants with onset at different ages and different types of inheritance. Usually manifested by muscle weakness of the hands and feet with a slow spread to the proximal muscle groups. Increased CPK levels; signs of myopathy on EMG and muscle biopsies.
Metabolic myopathies
They arise as a result of impaired muscle utilization of glucose and fatty acids as energy sources. Patients are diagnosed with either an acute syndrome of myalgia, myolysis and myoglobinuria, or chronically progressive muscle weakness. For an accurate diagnosis, it is necessary to conduct a biochemical and enzymatic study of muscle biopsies. However, muscle enzymes, EMG and muscle biopsies usually reveal pathological changes, and on their basis, a specific disease can be suspected.
In childhood and infant forms of glycogen storage disease, concomitant disorders are often identified with dysfunction of the heart, liver and endocrine system, the symptoms of which can complicate the diagnosis of muscle disorders. Forms found in children and adults can mimic muscular dystrophy or polymyositis. In some types of the disease, its manifestation may be one of the attacks of muscle cramps or muscle fatigue provoked by exercise. The lactate test for forearm ischemia has diagnostic value, since there is no increase in the level of lactic acid in the serum after exercise (characteristic of the norm). Disorders of fatty acid metabolism manifest themselves with clinical signs similar to those described above. In some patients, muscle cramps provoked by exercise, myolysis and myoglobinuria are detected; in others, the clinical picture resembles polymyositis or muscular dystrophy. Sometimes a special diet (including medium-chain fatty acids enriched in triglycerides), oral carnitine supplementation, or glucocorticoids are effective.
Other myopathies
Myopathies may be associated with endocrine disorders, especially with hypo- or hyperfunction of the thyroid gland, parathyroid glands and adrenal glands. Drugs (especially glucocorticoids) and certain toxins (including alcohol) are common causes of myopathy (Table 180-2); in most cases, muscle weakness is symmetrical and affects the proximal muscles of the limb girdles. Frequent symptoms: muscle weakness, myalgia, cramps. Diagnosis often depends on the interpretation of the signs of myopathy in the treatment of the underlying disease or elimination of the etiological agent, since laboratory studies of muscle enzymes, EMG, and even muscle biopsies do not provide specific data for diagnosis.
Diagnosis of congenital myopathy
Diagnosis of congenital myopathies is a complex process, since the doctor needs to differentiate and determine the specific type of myopathy in order to prescribe adequate treatment and make the correct diagnosis. A neuropathologist identifies neurological symptoms, conducts electrophysiological and biochemical studies to establish heterozygous carriage of the myopathic gene. Electromyographic examination using cutaneous electrodes often shows a decrease in the voltage of the EMG curve. When performing a biochemical blood test, an increased concentration of aldolase and creatine kinase is observed in the serum.
Diagnostics
- DNA diagnostics.
- Blood serum examination (increased CPK by 5–20 times, LDH).
- EMG (primary muscular nature of changes).
- Skeletal muscle biopsy (signs of primary muscular dystrophy and denervation).
- ECG/EchoCG (impaired atrioventricular and/or intraventricular conduction, myocardial hypertrophy, ventricular dilatation, congestive heart failure, cardiomyopathy).
Differential diagnosis:
- Progressive Duchenne muscular dystrophy.
- Erb's muscular dystrophy.
- Spinal amyotrophy Kugelberg-Welander.
- Polymyositis, dermatomyositis.
- Metabolic myopathy.
- Hereditary polyneuropathies.
Treatment of congenital myopathy
Treatment of congenital myopathies is ineffective. There is no clear treatment at the moment. Scientists are still arguing about whether congenital myopathy can be treated. Medical institutes in different countries are conducting research at the gene level using stem cells. There is symptomatic treatment, which consists of influencing metabolic processes in the patient’s body, in particular, protein synthesis, and an attempt to normalize the functions of the autonomic nervous system. Most often, drug treatment includes taking anabolic hormones (Nerobol, Ceraxon, Retabolil, Somazin), ATP. Vitamin therapy is mandatory. Anticholinesterase drugs are also prescribed.
A mandatory part of the treatment process for congenital myopathies is physical therapy. This could be exercise in water or a set of exercises. Exercise therapy is supervised by a trainer or neurologist. In some cases, orthopedic correction is effective (for example, wearing orthopedic shoes, corsets or the use of orthopedic mattresses, pillows, chairs).
The condition and clinical picture of the disease is monitored by a neurologist, therapist, pediatrician, cardiologist and orthopedist-traumatologist.
How to treat muscle dystrophy?
Hereditary disease is quite difficult to treat and correct.
But modern medicine offers treatments that help minimize the impact of the disease on the human body. The progression of the disease can lead to complications with the spine, leading to its deformation, as well as the development of breathing problems and frequent manifestations of pneumonia. Muscle failure affects the main human organ - the heart, because it is also a muscle.
The therapy can improve patients' quality of life by strengthening the heart, bone structure and lungs. Muscular dystrophy in adults and children is treated as follows:
- The patient uses corticosteroids to accumulate energy and reduce symptoms of the disease.
- The purpose of physical activity is therapeutic physical education, swimming, since the lack of muscle activity leads to faster progression of the disease.
- Physiotherapy is prescribed, the focus of which is maintaining muscle tone and improving the functioning of joints. The patient also uses special breathing exercises.
- The patient is prescribed the use of various orthopedic devices.
These measures may be poorly effective and may not help in resisting the development of the disease. If the respiratory system is compromised, the patient is supported to breathe using respiratory mechanisms.
Medicine is now developing, and new treatment methods are emerging that combat the progressive course of the disease. Such innovations include stem cell treatment, replacement or restoration of dystrophin (a protein that is responsible for maintaining the functions of skeletal and cardiac muscles), the creation of anti-inflammatory drugs with minimal side effects and drugs aimed at improving muscle functionality.
The latest innovation that provides an absolute cure for the disease is gene editing. This method is under development, and it is unknown when it will reach the stage of practical application.
Treatment and life prognosis
Treatment is symptomatic. Hormonal drugs are used to stop the destruction of muscle fiber, phospholipids to protect muscle cells from destruction, and elements of therapeutic exercises. Various orthopedic devices are being introduced into practice to facilitate movement. Massage is strictly contraindicated in most cases, as it can lead to accelerated muscle breakdown. Treatment of hereditary diseases is a matter of the future.
The life prognosis for patients is unfavorable. The course of the disease is progressive. Death is inevitable. As a rule, by the age of seven, severe symptoms develop, leading to complete immobility by the age of 13-14. Patients rarely live to 18-20 years of age.
Cause of the disease
Duchenne muscular dystrophy is based on a genetic defect of the sex X chromosome.
One of the sections of the X chromosome contains a gene that encodes the production of a special muscle protein called dystrophin in the body. The protein dystrophin forms the basis of muscle fibers (myofibrils) at the microscopic level. The function of dystrophin is to maintain the cellular skeleton and ensure the ability of myofibrils to undergo repeated acts of contraction and relaxation. In Duchenne muscular dystrophy, this protein is either absent altogether or is synthesized defectively. The level of normal dystrophin does not exceed 3%. This leads to the destruction of muscle fibers. Muscles degenerate and are replaced by fat and connective tissue. Naturally, in this case the motor component of human activity is lost.
The disease is inherited in a recessive manner linked to the X chromosome. What does this mean? Since all human genes are paired, that is, they duplicate each other, in order for pathological changes to appear in the body due to a hereditary disease, it is necessary that a genetic defect arise in one chromosome or similar sections of both chromosomes. If the disease occurs only with mutations in both chromosomes, then this type of inheritance is called recessive. When a genetic abnormality is detected in only one chromosome, but the disease still develops, this type of inheritance is called dominant. The recessive type is possible only when identical chromosomes are affected simultaneously. If the second chromosome is “healthy”, then the disease will not occur. That is why Duchenne muscular dystrophy is the lot of males, because they have one X chromosome in their genetic set, and the second (paired) Y chromosome. If a boy comes across a “broken” X chromosome, then he will definitely develop the disease, because the healthy chromosome he just doesn't have it. In order for Duchenne muscular dystrophy to occur in a girl, there must be a coincidence in her genotype of two pathological X chromosomes, which is practically unlikely (in this case, the girl’s father must be sick, and her mother must have a defective X chromosome in her genetic makeup). Girls act only as carriers of the disease and pass it on to their sons. Of course, some cases of the disease are not the result of inheritance, but occur sporadically. This means that a mutation appears spontaneously in the child’s genetic makeup. A newly appeared mutation can be inherited (provided the ability to reproduce is preserved).