Hippel-Lindau disease
(E. Hippel, German ophthalmologist, 1867-1939; A. V. Lindau, Swedish pathologist, 1892-1958; synonym:
multiple angioreticulomatosis, retinocerebrovisceral angiomatosis, familial angiomatosis
) - a disease characterized by the development of angiomatous, angioreticulomatous and cystic formations in the retina eyes, central nervous system and internal organs. Belongs to the group of phakomatoses (see).
Ophthalmological symptoms were first described by Panas (Ph. Panas, 1879) and Remy (A. Remy, 1892), and then in detail by Hippel in 1895, 1903, 1904. After classical studies by Lindau in 1926-1927, which proved the systemic nature of this disease and described cerebral disorders, Hippel-Lindau disease entered medicine as an independent nosological form. The disease is relatively rare.
Etiology and pathogenesis
The disease is hereditary. S. N. Davidenkov (1958), I. S. Babchin (1962), Shokeir (Shokeir, 1970) and others believe that angiomas of the retina, brain and hypernephroma have a single oncological genesis. Progression, the combination of angiomatosis with a tumor cell structure, and a familial nature are evidence of the uniform nature of the disease. In connection with the frequency of familial forms, one should speak not of a hereditary predisposition, but of a genetically determined disease with fairly stable gene penetrance and with insignificant influence of other genotypic factors and the environment. This is confirmed by the same age at the onset of the disease observed in several generations, the uniformity of forms, localization of angiomatous growths and combinations of certain anomalies in the development of the skeleton, endocrine system, and internal organs. The disease is inherited predominantly in an autosomal dominant manner.
Manifestations - oncological processes
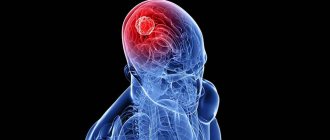
The main manifestations of Hippel-Lindau syndrome are tumors of the pituitary gland, cerebellum, spinal cord, retina, and internal organs. We list the possible neoplasms:
- Hemangioblastomas are localized in the cerebellum or retina; originate from nervous tissue. CNS neoplasms have an incidence of at least 60% in patients with Hippel-Lindau disease. The average age of patients diagnosed with this type of tumor of the central nervous system is 42 years.
- Angiomas are neoplasms of vascular origin localized in the spinal cord.
- Cysts. These are neoplasms that have a cavity filled with liquid inside. Such elements can be formed in the liver, kidneys, testicles and pancreas. In particular, cysts of the kidneys and pancreas occur in 33-54% of cases.
- Pheochromocytoma is a tumor arising from adrenal medulla cells. The frequency of occurrence is about 7% of cases. The average age of patients with pheochromocytoma is about 25 years.
- Cell carcinoma. In particular, the average age of patients with this tumor is 43 years.
- In men with Hippel-Lindau disease, benign neoplasms may develop in the testicular area (for example, papillary cystadenoma).
- In women, tumor lesions of the uterine ligaments occur.
Pathological anatomy
The pathological process of Hippel-Lindau disease is based on the development of capillary angiomas of the retina, followed by the formation of cysts, arteriovenous aneurysms with secondary proliferation of glia. In the retina, already in the initial period of the disease, clouding, dilation and tortuosity of blood vessels, mainly capillaries, are detected. Angiomas develop most often in the equatorial part, and primarily in the zone of capillaries between the arterial and venous trunks. Later, the arteries and veins become thickened, tortuous, dilated, the growing capillaries form glomeruli of a bright red color with a yellow tint, in which exudate and hemorrhages then appear. The progression of the process leads to retinal detachment (see), optic nerve atrophy (see). Histologically, cholesterol surrounded by lipid droplets (so-called pseudoxanthoma cells) is found in the protoplasm of tumor cells between the strands of capillaries. Angiomas, angioreticulomas, cystic formations, various anomalies of the ventricles and membranes are found in the brain. With the development of angioreticulomas with multifocal growth, multiple cysts and cavities are observed, and hemorrhages often occur. Various congenital anomalies of the development of internal organs are detected: cardiovascular system - coarctation of the aorta (see), congenital heart defects (see Congenital heart defects); polycystic kidneys, pancreas, liver. There are also pheochromocytomas (see), hypernephroma (see Adrenal glands, tumors).
The classical triad G.-L. b. are the above-listed vascular tumors and developmental anomalies of the retina, brain and spinal cord, and internal organs. However, the formation of tumor tissue in the body usually occurs unevenly. Incomplete forms of the disease are more common, when in some cases retinal angiomatosis predominates, in others the picture of intracranial angioreticuloma comes to the fore (see).
Therapy and prognosis of Hippel-Lindau disease
At the moment, the disease has only symptomatic treatment. Therapy is aimed at eliminating emerging malignant tumors. To detect tumors in a timely manner, you should be observed and examined by doctors every year.
The initial stages of retinal angiomatosis are considered a reason for focal radiation therapy, but after a year from the date of treatment there is a possibility of radiation retinopathy. For small angiomas, laser coagulation and diathermocoagulation can be performed; for large tumors, transscleral cryopexy can be performed. If you have central nervous system formations with Hippel-Lindau disease, you should visit a neurosurgeon.
It is possible to surgically remove tumors of the cerebellum, optic nerve and cerebral hemispheres. There are situations where stereotactic surgery has been used. If renal carcinoma is detected, then a partial nephrectomy is performed; if a pheochromocytoma is found, removal occurs. Surgical treatment of benign formations of the pancreas is possible if their growth is more than 2-3 cm.
The complete lack of treatment for the disease leads to blindness due to increasing angiomatosis of the retina, as well as fatal events due to an increase in formations of somatic and cerebral localization. With control and treatment of the disease, patients, according to statistics, live on average up to 40-50 years. About 50% of deaths are caused by hemangioblastomas of the central nervous system. At the initial stages, complete elimination of these formations is achieved in the majority of patients with Hippel-Lindau, but the neoplasms can recur, according to statistics, 6 years from the moment of their absolute removal.
Clinical picture
Members of the same family have various combinations of symptoms of G.-L.b. It begins between the ages of 10 and 30. Early signs are often ophthalmol. disorders manifested by a progressive decrease in vision and changes in the fundus. Then the symptoms of brain damage appear. With retinal angiomatosis, a peculiar picture of the fundus is observed. In the peripheral part of the retina, often in the lower segment, spherical reddish elevations are visible. They are the capillary angiomas described above. Several large looped feeding vessels approach them from the optic disc. Angiomas can be single or multiple and develop in one or both eyes. Both eyes are affected in 36-50%. During retinal angiomatosis, four stages are distinguished. In the initial stage, angiomas develop at the junction of several dilated arterial and venous branches, are small in size, and the retinal tissue is relatively preserved. In the second stage, the size of the angiomas becomes larger and they protrude into the vitreous body. Reactive gliosis develops, a sharp dilation of blood vessels, exudation, dystrophic changes and hemorrhages in the retina. In the third stage, exudation increases until the formation of segmental retinal detachment. In the fourth - terminal - stage, total retinal detachment occurs, a sharp degeneration of all structures of the eyeball.
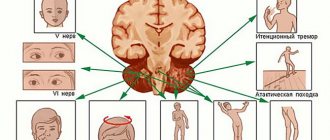
Neurol, symptoms of G.—L. b. depend on the localization of angioreticularis in one or another area of the brain. Most often, these tumors develop in the cerebellum, less often in the medulla oblongata, subcortical ganglia and in the cerebral hemispheres. Occasionally they are found in the spinal cord and nerve roots. Beginning of neurol, disorders with G.—L. b. It is more often observed between the ages of 20 and 40 years. When the angioreticuloma is located in the cerebellar hemisphere, general cerebral symptoms appear early, in particular, a periodically intensifying headache, especially in the morning, which can become permanent. Usually the headache is diffuse, but can be localized in the back of the head and radiate to the neck, back, and sometimes localized in the forehead. Headache is often accompanied by vomiting. Congestive optic discs and sometimes forced head posture are detected. Against this background, a disorder of statics and gait gradually develops, a picture of cerebellar ataxia (see), usually bilateral, but with a predominance on the side of the tumor, is formed.
With angioreticulomatosis of the medulla oblongata, early focal signs are vomiting, hiccups, dysphagia, cardiac and respiratory disorders, which are associated with the involvement of the nuclei of the IX and X pairs of cranial nerves in the process. Later, ataxia occurs, depending not only on damage to the posteromedial areas of the cerebellum, but also on the nuclei of the posterior funiculi. The disease develops over a number of years. General cerebral symptoms appear late.
With supratentorial localization of angioreticulomatosis, general cerebral symptoms appear first, but they proceed relatively mildly. Headaches occur in attacks, resembling a migraine. Epileptic seizures are observed, sometimes of the cortical type. The course of this form of G.—L. b. especially characterized by exacerbations (circulatory disorders in tumor tissue, manifested by increased cerebral and focal symptoms) with subsequent remissions.
Angioreticulomas of the spinal cord can cause radicular pain, loss of tendon reflexes and disorders of deep sensitivity (the result of posterior localization of the tumor in the spinal canal). Sometimes a picture of a transverse spinal lesion occurs. There is a combination G.-L. b. with syringomyelia (see), accompanied by the appearance of a corresponding symptom complex. Moderate protein-cell dissociation is detected in the cerebrospinal fluid; the pressure can be increased to 220, in some cases up to 330 mm water. Art. Angiography (see), pneumoencephalography (see Encephalography), scanning (see) help establish the location and sometimes the type of tumor.
Anomalies of development and neoplasms of internal organs in G.-L. b. develop latently and often remain unrecognized. Hypernephromas and pheochromocytomas developing from the adrenal gland cause an increase in blood pressure.
The course of Hippel-Lindau disease is slowly progressive. Remissions are sometimes observed. The disease, which begins in childhood, has a relatively benign course; it can become malignant at the age of 35-40 years and later. When angioreticularis are localized in the cerebral hemispheres, in the cerebellum, the progression of the disease, regardless of age, is extremely rapid. Features of the flow of G.-L. b. in childhood - the appearance of symptoms of damage to the nervous system against the background of existing ophthalmological changes. In some cases, the disease is complicated by iridocyclitis (see), secondary glaucoma (see), hemophthalmos (see), cataracts (see).
Diagnostics
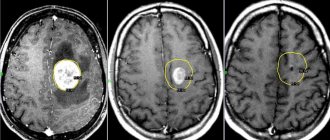
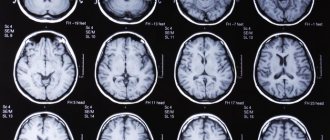
Only with timely consultation with a doctor can Hippel-Lindau syndrome be controlled. Thanks to modern developments in medicine and early diagnosis, the prognosis for patients has improved. Tumors can be detected during MRI, CT, ophthalmoscopy and other studies. A doctor may suspect the disease based on clinical symptoms. In this case, family history and the time when the disorders appeared must be taken into account.
A consultation with a neurologist is required to check the functioning of the cerebellum, an ophthalmologist to examine the retina, an oncologist to determine the type of tumor, as well as a geneticist. A genetic blood test is performed to detect the presence of a mutation in the gene responsible for the formation of tumors. If it is detected in one family member, the rest need to be examined regularly. And depending on the location of the formations, you will need to consult an endocrinologist, urologist and gastroenterologist.
In addition to MRI of the head, ophthalmoscopy and genetic analysis, to clarify the diagnosis, ultrasound of the kidneys, MRI of the spinal cord, ultrasound of the pancreas, CT scan of the adrenal glands, and retinal angiography are performed. A blood test for pancreatic enzymes and glucose is also needed. Typically, such studies are prescribed in all cases of retinal angiomatosis. They help differentiate Hippel-Lindau syndrome from retinopathy, neuropathy and retinoblastoma. In addition, regular examination of a child from the age of 10, whose close relatives have had kidney tumors, angiomas or hemangioblastomas, is mandatory.
Treatment
Previously used radiotherapy, x-ray irradiation, electrolysis, perforating diathermopuncture with diathermy in most cases did not provide sufficient effect. With the introduction of the photocoagulation method into ophthalmology (see Laser), reports appeared about the beneficial effect of xenon and laser radiation on angiomatous formations, especially in the early stages of the process. This allows us to consider photocoagulation as the method of choice. Treatment of angioreticulomatosis of the brain and spinal cord is surgical. In some cases, radiation therapy is used. Along with this, anticonvulsant, dehydration, restorative and restorative treatment is carried out according to indications.
Forecast
The prognosis depends on the form of the disease, in some cases it is unfavorable. If left untreated, the process progresses, leading to the death of the eyeball, rupture of angiomas, aneurysms, followed by hemorrhage in the brain. Timely surgical intervention eliminates brain disorders.
Bibliography
Arkhangelsky V. N. Morphological foundations of ophthalmoscopic diagnosis, p. 88, M., 1960; Katsnelson A. B. Retinal angiomatosis, Sov. Vestn. ophthalm., vol. 6, no. 4, p. 555, 1935, bibliogr.; Kotelyansky E. O. Intraocular tumors, p. 107, M., 1974, bibliogr.; Levkoeva E. F. Hippel-Lindau disease (retinal gliomatosis) in morphological light, in the book: Pathology of the retina and optic nerve, ed. K. V. Trutneva, p. 185, M., 1971; Merkulov I.I. Clinical ophthalmology, book. 2, p. 101, Kharkov, 1971; Shepkalova V. M., Khorasanyan-Tada A. A. and Disler O. N. Intraocular tumors, p. 211, M., 1965; Schmidt E.V. Angioretic in the brain, p. 136, M., 1955; Gahlot D. K., Khosia P. K. a. PrakashP. Retinocerebral angiomatosis, East. Arch. Ophthal., v. 2, p. 113, 1974; Hippel E. Uber eine sehr seltene Erkrankung der Netzhaut, Albrecht v. Graefes Arch. Ophthal., Bd 59, S. 83, 1904; Lindau A. Studien tiber Kleinhimcysten, Bau, Pathogenese und Beziehungen zur Angiomatosis retinae, Kobenhavn, 1926; Neurologie, hrsg. v. J. Quandt u. H. Sommer, Bd 2, S. 595, 803, Lpz., 1974; Rho JM Von Hippel - Lindau's disease, Canada. med. Ass. J., v. 101, p. 135, 1969; Trevor-Poper PD The eye and its disorders, p. 648, Oxford ao, 1974.
L. V. Kalinina, E. S. Libman.
Characteristic symptoms
Hippel Lindau's disease is characterized by many symptoms.
Thus, disturbances in the functioning of the cerebellum have the following manifestations:
- unsteadiness of gait – while walking a person may sway in different directions;
- lack of coordination of movements - a person misses when performing precise movements;
- tremor – there is trembling of the limbs;
- nystagmus - rhythmic oscillations of the eyeballs develop to the sides, and they are characterized by a fairly large amplitude of swing.
This disease is also characterized by increased intracranial pressure, which is associated with impaired outflow of cerebrospinal fluid from the brain.
This condition is characterized by the following manifestations:
- diffuse headache;
- nausea and vomiting;
- increased fatigue and weakness.
The disease is accompanied by the development of tumor formations in various organs and systems. This process is usually accompanied by the following manifestations:
- increased heart rate and bouts of heat in the body are characteristic of tumor formation in the adrenal medulla;
- swelling of the arms, legs, face, the appearance of blood in the urine - observed in pathological processes in the kidneys;
- infertility and pain in the testicles are characteristic of testicular tumors;
- diarrhea, a feeling of dry mouth and constant thirst - observed with a tumor of the pancreas;
- A decrease in visual acuity or its complete loss accompanies the appearance of a vascular tumor of the retina.