- 3 Июля, 2018
- Неврология
- Волощук Наталя
Заболевания нервной системы — не редкость. Они чаще всего проявляются невралгиями, но существуют и наследственные формы болезней, например, спиноцеребеллярная атаксия (СА). Неврологи используют термин «атаксия» для обозначения состояния, при котором наблюдается нарушение слаженности движений, контролирующихся сознанием. Иными словами, данный недуг представляет собой нарушение совместной деятельности мозжечка и спинного мозга. В чем проявляется недуг и как он лечится, рассмотрим ниже.
Описание
Атаксия спиноцеребеллярная представляет собой совокупность генетических недугов, которые носят неврологический характер, проявляются нарушением деятельности базальных ядер головного мозга и мозжечка и передаются по наследству. В результате этого изменяется координация движений и прочее. Негативным в заболевании является тот факт, что на сегодняшний день какого-либо определенного лечения не существует. Болезнь наследуется по механизму аутосомной доминантности, когда ферменты, которые появляются в результате мутаций генов структурных белков, деформируются. Обычно наследование происходит по отцовской линии из поколения в поколение.
Дополнительные симптомы
По мере развития спиноцеребеллярная атаксия обрастает новыми симптомами:
- возникает тремор конечностей после каких-либо действий или в результате нарушения координации;
- сильно меняется почерк – буквы становятся неровными, большими;
- постепенно изменяется речь, но этот признак сложно заметить, если жить с пациентом постоянно;
- возникают расстройства глазных движений – они становятся резкими, толчкообразными, когда человек перемещает взгляд с объекта на объект;
- в некоторых случаях ухудшается работа слухового аппарата;
- у многих людей нарушается стул, мочеиспускание;
- в запущенных случаях появляется паралич рук и ног;
- также заметно ухудшение привычных реакций и рефлексов, со временем они могут полностью исчезнуть.
У некоторых больных спиноцеребеллярная атаксия протекает в легкой форме, иногда переходит в стадию с присвоением временной инвалидности, но чаще всего не требует ее. Подобная картина встречается у людей, чьи близкие родственники страдают спиноцеребеллярной атаксией развернутого плана.
При спиноцеребеллярной атаксии всегда наблюдается потеря мышечной массы, которая в результате приводит к судорогам, выраженной мышечной слабости. Один из симптомов – ощущение подергивания, трепета под кожей.
Иногда при спиноцеребеллярном поражении возникают непроизвольные повороты и подергивания головой, развивается слабоумие, признаки которого похожи на болезнь Паркинсона. Для нарушения характерны и проблемы со зрением. Тяжелая форма сопровождается полной утратой зрительного контакта.
Классификация
Спиноцеребеллярная атаксия (мкб 10 — G11) в 90% случаев представляет собой шесть из двадцати генетических вариантов болезни. Данные варианты были классифицированы по номерам: 1, 2, 3, 6, 7 и 8 типы атаксии. Патология выражается в изменении количества CAG в части генов, которые кодируют больные гены. Рассмотрим далее эти типы подробнее:
- Атаксия первого типа сегодня самая распространенная. Она возникает вследствие размещения в шестой хромосоме мутированного гена ATXN1. В норме этот ген имеет тридцать шесть повторов, при большем же их количестве развивается болезнь. Мутация гена вызывает образование ДНК белка, который принимает участие в метаболизме клеток. Это способствует дегенерации и развитию болезни.
- СА второго типа распространена несколько меньше. Она характеризуется увеличением повторов в двенадцатой хромосоме. Какую функцию при этом выполняет белок, медицине неизвестно.
- Третий тип именуется болезнью Мачадо-Джозефа. В этом случае нарушение происходит в гене, что размещен в четырнадцатой хромосоме. Белок при этом принимает участие в обмене энергией между мозжечком и ядрами мозга.
- Спиноцеребеллярная атаксия шестого типа является редким недугом. Здесь происходит нарушение в гене, который находится в девятнадцатой хромосоме. Ген кодирует белок, который размещается в нейронах мозжечка. Этот процесс вызывает также наследственную форму мигрени.
- СА седьмого типа обуславливается нарушениями гена в третьей хромосоме. Какую функцию выполняет белок, медицине неизвестно.
- Восьмой тип характеризуется изменениями гена в тринадцатой хромосоме.
Наследственные формы генетических отклонений
По частоте возникновения спиноцеребеллярная патология занимает 2 место среди всех заболеваний ЦНС. Единой классификации патологий нет, так как симптомы у многих видов одинаковы, врачи не могут точно делить патологию на разные категории. Существует 3 распространенных типа по симптомам и причинам. Но есть деление на формы согласно повреждению генов (всего их 8).
Болезнь Фридрейха
Среди 120 человек случайно выборки 1 оказывается носителем этого гена. Передается он и мальчикам, и девочкам. Развивается в результате следующих причин:
- нарушение синтеза белка, отвечающего за транспорт ионов железа;
- уничтожение клеток ЦНС, миокарда, а также островков поджелудочной железы;
- на поздней стадии страдают ножки мозжечка, ядра нервов.
Первые симптомы спиноцеребеллярная атаксия вызывает у детей в 10-11 лет: изменения походки, пошатывания, сложности передвижения в темноте. Постепенно меняется координация движений, портится почерк. Затем наступают классически симптомы – атрофия мышц и утрата подвижности.
Неврологи у больных больше не наблюдают сухожильных рефлексов. Постепенно в дистрофический процесс вовлекаются остальные органы. Происходит деформация столба позвоночника, пальцев рук и ног. У пациентов нередко наблюдается сахарный диабет, проблемы с яичниками, замедляется половое созревание, возникает ожирение.
Атипичная форма болезни развивается после 30-50 лет, протекает легче, чем в детском возрасте. За счет медленного прогрессирования лучше поддается коррекции.
Атаксия с нехваткой витамина Е
Наименее распространенная форма спиноцеребеллярной атаксии, при которой ухудшается внедрение витамина в структуру белков. В результате сильно страдает антиоксидантная защита организма, возникают симптомы, аналогичные болезни Фридрейха.
Отличается спиноцеребеллярная атаксия при недостатке витамина Е симптомами со стороны сердца, костной и эндокринной системы – они практически не страдают. Больше всего угнетаются рефлексы и речевые возможности человека. К 30 годам пациент почти полностью утрачивает возможность самообслуживания.
Аутосомно-доминантные формы
В эту категорию входит 13 самостоятельных заболеваний, мало отличающихся друг от друга по симптомам. К ним приводят мутации 13 генов.
Спиноцеребеллярная атаксия каждого типа вызывает гибель определенных клеток нервной системы:
- Диагностируется в основном от 30 до 40 лет. Первый тип сопровождается нарушениями походки, изменением координации и мышечного тонуса. Постепенно развивается деменция, тазовые болезни, треморы, двигательные проблемы.
- Второй тип спиноцеребеллярной патологии дополняется глазными расстройствами, но в целом похож на первый.
- Третий тип – болезнь Мачадо-Джозефа появляется при поражении мозжечка. Главные отличия от других типов: насильственные движения, подергивания лицевых мышц.
- Четвертый тип сопровождается острой болью и нарушениями чувствительности в той области, за которую отвечает поврежденный нерв.
- Спиноцеребеллярная патология 5 и 6 типа появляется уже после 50 лет, прогрессируют медленно и сохраняют уровень жизни пациента.
- Седьмая форма сопровождается двигательными расстройствами, связанными с глазами, нередко наблюдается слепота.
- Восьмой тип и последующие формы болезни плохо изучены, встречаются крайне редко (менее 1 случая на 500 000 человек).
Спиноцеребеллярная атаксия – тяжелое неврологическое расстройство, которое не поддается полному излечению и сопровождается острыми симптомами по мере развития. Однако спиноцеребеллярное наршуение можно замедлить, если вовремя его обнаружить.
Причины
При любом типе заболевания происходит мутация гена, приводящая к образованию ДНК белка патологической формы, который богат глутамином. Он вызывает появление в ядрах нейронов мозжечка и базальных ядер мозга отложений в виде агрегатов, нарушая свойства протеинов. Белки принимают участие в обмене веществ, протекающих в нервной ткани. Скорость протекания данного процесса зависит от количества поворотов в гене, которое отличается от нормы. Это определяет симптоматику заболевания. При созревании половых клеток симптомы усиливаются.
Симптомы
Все типы данного заболевания имеют одинаковую симптоматику, различными могут быть только второстепенные элементы. Так, спиноцеребеллярной атаксии симптомы не проявляются в детском возрасте. Средний возраст людей, страдающих недугом, составляет от восемнадцати до тридцати лет. Атаксия третьего, шестого и седьмого типов развивается позже, обычно это происходит после тридцати лет. Первым признаком недуга является появление неуклюжести при ходьбе и беге. Позже наблюдается тремор конечностей, изменение походки, офтальмоплегия, почерк меняется. Со временем недуг приводит к развитию паркинсонизма. При некоторых видах болезни наблюдается атрофия зрительного нерва. СА шестого, седьмого и восьмого типов характеризуется нарушением речи и процесса глотания, что приводит к истощению. Истощение вместе с патологиями часто провоцируют смертельный исход. При всех видах заболевания нарушается координация движений. Средняя продолжительность жизни больных составляет от десяти до двадцати пяти лет, в зависимости от формы недуга и качества ухода за ними.
Лечение в Италии
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Спиноцеребеллярная атаксия — лечение в Италии
Спиноцеребеллярная атаксия включает в себя различные активно развивающиеся наследственные формы патологий, характеризующиеся нарушением координации движений. При таких поражениях организма человека основные изменения происходят в мозжечке, в спинном мозге, а также в стволе головного мозга. Среди всех известных наследственных болезней спиноцеребеллярные атаксии находятся на втором месте по частоте возникновения после нервно-мышечных патологий.
Заболевание отличается заметным клиническим многообразием симптомов, есть широкая градация между смешанными формами патологии и мозжечковыми типами. Известно множество различных классификаций, основанных на клинико-анатомическом принципе и на типах генетического наследования признаков заболевания.
Важно знать, что все известные классификации спиноцеребеллярных дегенеративных процессов не в состоянии полноценно удовлетворить требования медицины. В основном это можно объяснить недостаточным изучением болезни и отсутствием основных представлений о развитии биохимических дефектов, которые становятся причинными.
Спиноцеребральная атаксия встречается с частотой в разных популяциях приблизительно в 1 — 23 случаях на сто тысяч населения. Патология обладает неравномерным этническим и географическим распространением. В России и в Италии в основном преобладает разновидность первого типа болезни, в Индии — чаще встречается второй тип спиноцеребеллярной атаксии, заболевание третьего типа чаще всего диагностируется у населения Германии, США и Японии.
Группа спиноцеребеллярных атаксий относится к повреждениям мозжечка, которые наследуются по аутосомно-доминантному типу. При этом происходящие в генах мутации провоцируют формирование патологических белковых продуктов, вызывающих отмирание и гибель клеток в мозжечке, спинном мозге, в коре полушарий головного мозга.
Неврологические симптомы спиноцеребеллярной атаксии развиваются довольно медленно, а само по себе развитие может затягиваться на срок до двадцати лет, но может встречать и более быстрое прогрессирование заболевания. Порой наблюдаются периоды стабильного состояния.
При развитии сопутствующих инфекционных поражений начинают проявляться дополнительные симптомы. Пациенты с сильно запущенными стадиями патологии лишаются возможности вставать с постели, у них формируется дисфагия, то есть нарушение акта глотания, и другие подобные признаки. Человек может умереть в связи с истощением и часто от развития миокардита, сопровождающегося тяжелыми формами недостаточности сердца. При надлежащем уходе больной доживает до сорока — пятидесяти лет.
Впоследствии симптоматика дополняется тремором ног и рук при реализации каких-либо действий и при нарушении координации движений. Кроме того, начинается изменение почерка — он становится неровным, а буквы слишком большими, также изменяется речь.
Для заболевания характерны и расстройства движения глаз — толчкообразные, резкие движения глазных яблок при смещении взгляда с одного объекта на другой. Часто отмечается возникновение нарушений глотания, произношения речи, нарушения работы слухового аппарата, нарушение стула и отхождения мочи, паралич ног и рук, развиваются патологические рефлексы. А привычные рефлексы одновременно ухудшают реакции и затем исчезают полностью.
Временами заболевания протекает в абортивной или легкой форме и провоцирует незначительную инвалидность пациента или вовсе не сопровождается инвалидностью. Такие патологии могут диагностироваться у родственников больного, которые страдают развернутыми клиническими типами болезни.
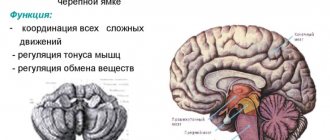
При диагностике предпочтение отдается методам визуализации, таким, как компьютерная или магнитно-резонансная томография. Они позволяют определить участки дегенерации нервных волокон, демиелинизацию нейронов моста, расширение всех компонентов системы циркуляции ликвора в головном мозгу, атрофические изменения коры полушарий головного мозга.
Для постановки диагноза также необходимо исключить и некоторые другие заболевания, протекающие со сходной клиникой (такие, как опухоли задней черепной ямки, рассеянный склероз, гидроцефалией, сосудистой патологией головного мозга, особенно в позвоночно-базиллярном отделе. Если на основании клиники удается подтвердить развитие атаксии, то гораздо более сложным является определение типа заболевания (если нет строго специфичных симптомов).
В настоящее время существует несколько мнений относительно тактики лечения данной патологии. Сторонники первой придерживаются мнения, что атаксия не подвержена медикаментозному лечению. В таком случае, лучше всего проводить поддерживающую терапию, способствующую замедлению прогрессирования заболевания. В обязательный комплекс входят ЛФК (вестибулярные тренировки), а также многие методы социальной, бытовой и трудовой реабилитации.
Другая же теория предполагает использование некоторых препаратов, однако, лечение проводится сугубо симптоматическое или стимулирующее. Назначаются инъекции витаминов, а также их пероральный прием. Как элемент такой терапии, возможно назначение противосудорожных препаратов (по показаниям). Лечение спиноцеребеллярной атаксии поддерживается и некоторыми физиотерапевтическими процедурами (например, электростимуляция).
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 – срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Диагностика
Прежде всего, проводят неврологический осмотр больного, изучают анамнез, проводят МРТ и различные молекулярно-генетические анализы и ДНК-диагностики. На разных стадиях развития недуга врач выявляет разные нарушения, связанные с неврологией. Это может быть тремор, нарушение речи, дисфагия и прочее. Некоторые формы заболевания обуславливаются быстрым развитием зрительных нарушений, что приводят к полной слепоте. Заболевание склонно прогрессировать. При исследовании наследственного анамнеза может быть обнаружено аутосомно-доминантное наследование от отца. МРТ показывает нарушения в области больших полушарий, мозжечка и базальных ядер. Может также наблюдаться атрофия мозжечка. Молекулярное исследование и ДНК-диагностика указывают на увеличенное число повторов в генах больного.
Генный сбой
Данное заболевание является наследственным и развивается вследствие того или иного генетического сбоя.
При этом, оно может проявляться в двух формах – с нарушением устойчивости и поддержания тела в пространстве (статико-локомоторная атаксия с поражением червя мозжечка) или же с поражением полушарий мозжечка и нарушением совершаемых конечностями активных движений (динамическая форма церебеллярной атаксии).
Заболевание наследуется по аутосомно-доминантному типу. Первые признаки проявляются обычно в возрасте старше 30 лет.
Довольно редко клиника может развиться и раньше, и тогда она, чаще всего, проявляет себя параллельно с иными неврологическими симптомами.
Благодаря изучению геномного состава было доказано, что в развитии спиноцеребеллярных атаксий ведущая роль принадлежит генам.
Доказано, что существует, как минимум, 13 специфических генов, отвечающих за развитие атаксии, а конкретнее – определенного его типа.
Каждый из этих генов находится в определенной хромосоме, а ее поражение в результате действия мутагенов и приводит к развитию заболевания.
В настоящее время известно 8 типов рассстройства. Спиноцеребеллярная атаксия 1-13 типов зависят от поражения одного определенного гена.
Основная роль принадлежит увеличению количества триплетов ЦАГ (цитозин-аденин-гуанин). В норме, данной последовательностью нуклеотидов кодируется образование глутамина – аминокислоты, необходимой для образования связей между белками головного мозга.
Из-за увеличения ее количества происходит удлинение белковых цепей, в результате чего начинают образовываться нерастворимые компоненты, из-за которых нарушается взаимодействие между отделами головного мозга и происходит гибель некоторого количества нервных клеток, что и способствует развитию атаксии.
Если рассмотреть вещество мозжечка, то можно заметить, что его полушария подверглись дегенеративным процессам (а также и основные компоненты, участвующие в проведении нервного импульса от спинного мозга к мозжечку и обратно (например, оливы)).
Параллельно с этим наблюдается демиелинизация нервных волокон, что препятствует быстрому распространению нервного импульса.
Статья в тему: Какие успокоительные средства для нервной системы лучше и эффективнее?
Лечение
Какого-либо эффективного лечения данного недуга на сегодняшний день нет. Результативность поддерживающей терапии на данный момент не доказана, однако она проводится для замедления развития недуга. Так, спиноцеребеллярная атаксия лечение предполагает в виде витаминотерапии и средств, которые стимулируют обмен веществ и метаболизм в нервной ткани. Также больным назначают ноотропные препараты. Немаловажную роль играет и физическая культура. Врачи рекомендуют больным выполнять комплекс упражнений для укрепления мышц и уменьшения нарушений равновесия. Проводят сеансы массажа и электромиостимуляцию.
Возможно ли излечение
На данный момент имеются несколько подходов осуществления лечебных мер для этой болезни.
Одни исследователи утверждают то, что медикаментозные препараты бессильны против атаксии, лучший эффект дадут поддерживающие терапевтические меры, замедляющие болезнь. Комплексное лечение состоит из лечебной физкультуры (вестибулярных тренировок), с другими социальными, бытовыми реабилитационными мерами.
Другие считают, что надо использовать некоторые средства для симптоматического или стимулирующего лечения.
Как правило, применяются витаминные препараты, которые вводят инъекционно либо принимают перорально. Согласно показаний, лечащий врач назначит противосудорожные средства.
Не лишним будет проведение физиотерапевтических процедур, например, электростимуляционного воздействия, выполнение классического массажа.
Прогноз
Как правило, прогноз при данном заболевании неблагоприятный, поскольку недуг постоянно прогрессирует и приводит к инвалидизации, а затем и к летальному исходу. Поэтому ответ на вопрос о том, лечится ли спиноцеребеллярная атаксия, будет отрицательным. Данное наследственное заболевание неизлечимо. В некоторых случаях прогноз может быть не столь негативным. Это бывает при развитии заболевания в преклонном возрасте и своевременном лечении, тогда большое количество симптомов могут не проявиться. Если недуг обнаружился в молодом возрасте, длительность жизни таких пациентов будет невелика. Больные с пятым и шестым типом недуга живут нормальной жизнью немного дольше, обычно срок их жизни не меняется. При правильном уходе и своевременной терапии врачам удается увеличить время жизни больных на десять лет. В среднем с таким заболеванием, как спиноцеребеллярная атаксия, живут около двадцати лет. Причиной летального исхода часто становится сердечная недостаточность и наличие инфекций.
Поддается ли болезнь лечению?
Способов лечения спиноцеребеллярной атаксии существует несколько, и не все они подтверждают возможность терапии медикаментами. Некоторые медики уверены, что лучший способ – это поддерживающая терапия с применением ЛФК, а также методов социально-бытовой реабилитации. Спиноцеребеллярные нарушения, согласно этой технологии, можно только замедлить, но избавить от нее пациента полностью не получится.
Второе направление терапии – применение препаратов для устранения симптомов болезни, а также медикаментов, стимулирующих работу организма. В рамках этой технологии назначают витамины в разных формах, а также противосудорожные препараты, если есть симптомы. Физиотерапевтические методы типа УВЧ и электрофореза могут использоваться, и эффективность этих способов достаточно высока.
Длительность жизни при диагнозе редко превышает 20 лет. Но если вовремя начать поддерживающую терапию, то можно увеличить этот показатель на 5-7 лет минимум.
Наихудший прогноз ждет людей, у которых патологию нашли раньше 20 лет. Прогресс заболевания в этом случае стремительный – в течение 2-3 лет. А люди со спиноцеребеллярной атаксией 5 и 6 типа живут дольше, процесс их восстановления проходит легче, а симптомы не так выражены.
Профилактика
Профилактические меры представляют собой медицинское и генетическое консультирование родителей, в чьем анамнезе наблюдались такие состояния. Также проводится генетическая перинатальная диагностика. Врачом определяется риск появления недуга у прямых родственников. Риск развития патологии для здоровых братьев и сестер, а также детей больного составляет 50%. В свою очередь, дети этих людей имеют вероятность унаследовать болезнь в 25%. Все эти лица находятся в группе риска и представляют собой главные объекты для консультирования. Основой профилактического исследования является ДНК-диагностика лиц из группы риска на наличие мутированных генов.
Спиноцеребеллярная атаксия в современное время является тем заболеванием, которое не лечится и приводит со временем к летальному исходу. Предупредить развитие заболевания можно при помощи специальных методов диагностики, но предотвратить его развитие невозможно, поскольку недуг этот имеет наследственный характер и обуславливается мутациями здоровых генов. Все это подталкивает современную медицину к разработке методов исследования на самых ранних этапах развития болезни, а также изучению причин мутаций, которые передаются по наследству от отца к детям.
Прогноз неутешительный
Что касается прогноза заболевания, то больные с атаксией живут в среднем около 15 – 20 лет. При надлежащем уходе, а также своевременно диагностированном заболевании и использования поддерживающей терапии, удается продлить жизнь на 7-10 лет.
Пациенты с атаксией 5 или 6 типов могут жить нормально несколько дольше (срок жизни обычно сильно не изменяется).
Наихудший прогноз для лиц, у которых заболевание развилось в раннем возрасте (обычно, возможно стремительное прогрессирование на протяжении пары лет).
Учитывая, что заболевание плохо подвергается лечению, необходимо совершенствовать имеющиеся методы диагностики, а также пытаться открывать и внедрять новые тактики лечения.