About the disease
First you need to figure out what it is.
Thomsen's myotonia is a chronic neuromuscular disease with a slowly progressive course.
Some people confuse the doctor's name and mistakenly believe that the disease is named after a certain Thompson, but this is incorrect.
The disease is a congenital genetic disorder and is inherited in an autosomal dominant manner. The detection rate is 0.3-0.7% per 100,000 newborns. It manifests itself as a slow relaxation of striated muscles in combination with a tonic spasm following the onset of voluntary movement.
ICD-10 code: G71.1.
How to identify pathology?
The diagnosis can be easily determined by external signs. It is important to carefully collect family history and clinical features.
At the first appointment, the specialist uses a neurological hammer. By lightly tapping problem muscle areas, the neurologist determines the ability of the muscles to contract and records the time of relaxation after irritation. If a ridge forms at the point of contact, this indicates a myotonic symptom.
The doctor asks the patient to clench his fingers into a fist and try to unclench them. The first movements may be difficult, and then normalization of motor skills occurs, which means that tonic spasms are present.
Muscle tissue is in good shape even at rest, and tendon reflexes exhibit myotonic signs.
The tonic symptom complex is characteristic not only of Thomsen’s myotonia. It accompanies Eulenburg's paramyotonia, Becker's myotonia, Steiner's myotonia, as well as other neuromuscular and endocrine disorders. Differentiation of diagnosis and designation of a specific type of myotonia are quite difficult when diagnosing.
Etiology and pathogenesis
The development of the disease is associated with a genetic mutation that develops during the prenatal period. The damaged gene responsible for the operation of chloride channels and the synthesis of the dystrophin protein is located on the long arm of chromosome 7 . As a result of the mutation, insufficient formation or complete absence of the protein that regulates muscle contraction occurs.
Normally, dystrophin is responsible for maintaining muscle function and the correct sequence of relaxation and contraction. Due to disruption of its synthesis, chlorine is retained on the membranes of muscle cells. The striated muscle bundles lose their ability to relax and remain contracted after performing the movement.
Overvoltage of the fibers causes an even greater accumulation of chloride ions on the surface of cell membranes. The balance between the remaining ion channels is disrupted. Accumulated chlorine provokes the release of calcium ions outside the cells. Excess calcium has a direct damaging effect and leads to the destruction of muscle cells.
REFERENCE. Tonic spasms gradually cause deformation of the entire axial skeleton. Due to damage to the respiratory muscles, episodes of shortness of breath or suffocation develop. Damage to the striated muscle of the heart leads to cardiomyopathies.
Symptomatic manifestations
The defining symptom in the clinical manifestation is myotonic syndrome. A condition in which the muscle receives a tonic spasm and is held in this position for 5 to 30 seconds.
The deviation is especially noticeable during rapid movement with applied force. At the first attempts to resume motor activity, the patient experiences serious difficulties. Repeated efforts are more effective: the rate of muscle relaxation stabilizes, movements return in full, almost like those of a healthy person. But keeping muscles at rest again leads to a similar problem - loss of normal muscle relaxation.
The most pronounced contractures and spasms are in the hands, chewing and facial muscles. When speaking, there is a disorder of speech activity and difficulties with articulation. Eating solid foods and swallowing are difficult. In severe cases, the disease is complicated by simultaneous stiffness of many muscle groups. As a result, when the movement starts abruptly, balance is lost, which leads to a fall and temporary immobility. Stressful situations, the influence of low temperatures, and heavy physical activity speed up myotonic reactions.
People suffering from Thompson's myotonia have an athletic build. At the same time, the appearance absolutely does not correspond to the true muscle strength, which is reduced due to the formed hypertrophy.
Classification
The disease is divided into two clinical forms: generalized and localized .
The generalized form is damage to the entire skeletal muscles of the body. With this form, hypertonicity of the facial muscles may or may not be observed.
Localized form - damage to one muscle group. Observe the development of myotonia:
- Facial muscles – bilateral, unilateral;
- Language;
- Chewing muscles;
- Circular muscles of the eye ;
- Shoulder girdle and upper limb ;
- Torso muscles;
- Muscles of the pelvis and lower limbs.
IMPORTANT: One form of the disease never develops into a second, which indicates the initially specified nature of the lesion.
Classification of myotonia
According to the common classification, the following types of myotonia are distinguished:
- congenital myotonia, known as Thompson's disease;
- cold paramyotonia (Eilenburg disease);
- atrophic myotonia (Rossolimo-Batten-Steinert-Kurshman disease; dystrophic myotonia);
- paradoxical myotonia;
- Schwartz-Jampel syndrome.
In addition, there is a classification based on the etiological factors that trigger the development of the myotonic phenomenon. Based on this classification, the following are distinguished:
- percussion myotonia;
- action myotonia;
- electromyographic myotonia.
Patients with percussion syndrome experience muscle contractions when struck vigorously with a hammer. With myotonia of action, the patient feels difficulty when trying to quickly clench and unclench a fist. In the case of electromyographic myotonia, when performing an electromyogram, discharges are detected, the frequency of which first increases and then decreases. In this situation, the phenomenon of myotonia is caused by worsening membrane instability in the muscle fiber structure.
Clinic of the disease
The main symptoms are myotonic spasm and muscle stiffness:
- Myotonic spasm is a prolonged muscle contraction following an active movement, lasting several tens of seconds. The patient is unable to eliminate the spasm voluntarily;
- Muscle stiffness is difficulty performing movements after a period of rest. Stiffness goes away after the patient tries to perform the movement several times.
The first manifestations of the disease are observed in the infancy period. While crying, the baby suddenly begins to choke and his voice changes. After calming down, the grimace remains on the face for 1-2 minutes.
Already in childhood, due to muscle hypertrophy, children resemble professional athletes, but muscle strength is significantly reduced. During physical activities, running, swimming, children are significantly inferior to their peers, despite their appearance.
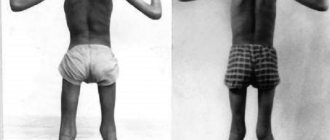
REFERENCE. The disease is not accompanied by pain and progresses during the cold season. Muscle stiffness often affects patients after sleep and leads to gait disturbances (symptoms of “tin man”, “duck walk”, “climbing stairs”).
In the generalized form, due to contraction of several muscle groups, patients lose their balance. A fall is accompanied by the inability to get up on your own until the muscle spasm disappears.
In the localized form, the following tonic contractions are observed:
- Hand muscles - during writing and performing small movements;
- Foot muscles - during walking and physical exercise;
- Language – during a conversation;
- Eye muscles - during squinting;
- Masticatory muscles - during chewing, talking.
Muscle hypertrophy even in adulthood makes patients “professional athletes,” but their physical fitness is significantly lower than that of their peers. Everyday and professional movements, as well as walking, are difficult and sometimes impossible. The patient cannot quickly unclench his hands, open his eyes, mouth, or change position.
Myotonia
Myotonia is a hereditary disease related to channelopathies (diseases associated with pathology of ion channels). Manifested by delayed muscle relaxation. Characteristic signs of myotonia are myotonic discharges, detected by needle EMG, and myotonic phenomena, which are detected during clinical examination. Congenital myotonia is accompanied by muscle hypertrophy; dystrophic myotonia, on the contrary, is accompanied by muscle atrophy. Diagnosis of myotonia is carried out using EMG, ENG and evoked potential studies. To date, radical drug therapy for myotonia has not been developed. Patients receive symptomatic and metabolic treatment, massage, exercise therapy, and electrical stimulation. Clinical picture of myotonia
Pathological impairment of muscle relaxation during myotonia is caused by impaired permeability of cell membranes, changes in ion and mediator exchange. In atrophic myotonia, clinical symptoms are explained by dysregulation of the hypothalamic-pituitary system or the pleiotropic effect of a mutagenic gene.
The 'fist' symptom is the main clinical test for identifying myotonia: the patient cannot quickly unclench his fist, for this he needs time and some effort. With repeated attempts, this myotonic phenomenon fades away with the exception of Eulenburg myotonia, when stiffness, on the contrary, intensifies with each repeated attempt. Stiffness is also observed when unclenching clenched jaws, quickly opening closed eyes, quickly getting up from a chair. Needle electromyography reveals one of the most characteristic phenomena for myotonia—myotonic discharges accompanied by the sound of a “dive bomber” that occur when a needle electrode is inserted and moved.
A distinctive clinical feature of congenital myotonia is the hypertrophy of individual muscle groups, which creates the impression of the patient’s athletic physique. In most cases, muscle strength is preserved, but sometimes it is reduced in the distal muscles of the arms. Dystrophic myotonia is a multisystem disease. In most cases, neurological symptoms are combined with cardiac pathology (left ventricular hypertrophy, arrhythmia), cerebral symptoms (hypersomnia, decreased level of intelligence), endocrine disorders (menstrual irregularities in women; hypogonadism and impotence in men). For paramyotonia, the so-called 'cold myotonia' - the occurrence of muscle spasm and paresis in the cold; such attacks can last from several minutes to several hours. Clinical manifestations of neuromyotonia include muscle stiffness, spasms (painless), and persistent muscle activity on the EMG.
Treatment of myotonia
The goal of treatment of neuromyotonia is to eliminate constant muscle activity and achieve possible remission; the goal of treatment of myotonia is to reduce the severity of myotonic manifestations. Non-drug treatment of myotonia consists of a diet with a restriction of potassium salts, exercise therapy, massage, electrical myostimulation, as well as prevention of hypothermia, since all myotonic reactions intensify in the cold. There is no radical drug treatment for myotonia, therefore, in order to reduce the severity of myotonic manifestations, phenytoin (200-400 mg/day) is used, and diuretics are used to reduce potassium levels. In some cases, it is possible to achieve remission with the help of immunosuppressive therapy: intravenous administration of human immunoglobulin (400 mg/kg), prednisolone (1 mg/kg/day), cyclophosphamide. In severe cases, a short course of treatment with glucocorticoids is carried out, calcium antagonists and diuretics (diacarb) are used. For atrophic myotonia, along with the indicated therapy, anabolic hormones are indicated in combination with fractional transfusions of blood, plasma, vitamin E, B complex vitamins, and small doses of ATP.
Becker's myotonia
This is a congenital neurological disease from the myotonia group, inherited in an autosomal recessive manner. Refers to sex-linked diseases (caused by a genetic mutation in the X chromosome region). It has a late onset, a milder course and slower progression. Unlike Thomsen's disease, clinical manifestations typically begin at 8-10 years of age. The clinical picture for both diseases is identical, but with Becker myotonia the manifestations are less pronounced. Both diseases are classified as hypertrophic myotonia.
| Becker | Thomsen | |
| Location of mutation | X chromosome | Seventh chromosome |
| Age of onset | 8-10 years | Breast period |
| Inheritance | Recessive (gene mutation was present in both parents) | Dominant (mutation was present in at least one parent) |
| Clinical picture | Less pronounced | Bright |
Diagnostic criteria
A doctor may suspect Rossolimo-Steinert-Kurshman disease if the patient has a combination of myotonic and dystrophic changes in the muscles against the background of loss of intelligence and the presence of cardiovascular and endocrine pathology.
Polysystemicity almost always indicates the genetic nature of the disease. Such patients are subject to DNA analysis and genealogical analysis to confirm the autosomal dominant inheritance of the pathology. Electrocardiography, electroneuromyography, and hormone tests are used as informative research methods.
Due to the versatility of clinical manifestations, specialists from various branches of medicine are usually involved in the diagnosis process - genetics, cardiology, endocrinology, gynecology, andrology, neurology.
Differential diagnosis is made between dystrophic myotonia and other types of similar diseases. Unlike the others, Rossolimo's disease is characterized by muscle atrophy. Often, to confirm the diagnosis, it is necessary to resort to a biopsy to determine the level of muscle protein, which is increased in the tissues of this pathology.
Antenatal diagnosis is also carried out by examining amniotic fluid.
Dystrophic myotonia Rossolimo-Steinert-Kurshman
This type of myotonia is also hereditary. This disease is progressive and is based on a defect in myotonin protein kinase. This leads to dystrophic changes in muscle tissue.
The classic form of this disease appears between the ages of 10 and 20 years. In rare cases, this myotonia is congenital. In this case, the disease is already visible at birth. With Rossolimo-Steinert-Kurshman myotonia, replacement of part of the muscle fibers with adipose and connective tissue is observed.
Symptoms of Thomsen's myotonia
Thomsen's myotonia is a genetic disease, but external clinical signs are not detected immediately after birth. In most cases, the first symptoms appear in childhood (5-8 years) and adolescence (up to 20 years).
A feature of the clinical picture is the myotonic phenomenon, characterized by:
- muscle hypotonia at rest;
- hypertonicity, spasm of muscle fibers at the moment of volitional efforts;
- prolonged muscle relaxation after the start of active movement.
Depending on the area of damage, myotonic attacks can affect the legs, arms, muscles of the neck, shoulders, and face. As a result, muscle spasms are observed when you want to spread your fingers clenched into a fist, when you start walking, when you close your jaws, close your eyes, etc.
If Thomsen's myotonia manifests at an early age, signs of muscle weakness are determined by the difficulty of the child's physical development. The baby does not sit, does not get up, does not walk in the prescribed time, his body becomes uncontrollable.
At a later age, myotonic attacks of skeletal muscles appear when walking, the desire to stand up, maintain balance, that is, with any voluntary movement. The patient, performing the first motor act, feels a sharp muscle spasm and becomes immobilized. Wanting to get up, a person suffering from this disease must lean on something. The first step when moving is given with great difficulty, sometimes the tonic spasms are so strong that the patient falls. When clenching your fingers into a fist, it is impossible to unclench them for 10 seconds or more, even if maximum effort is applied. Subsequent movements are easier and spasms stop.
During vigorous activity, the affected muscles adapt to movement and spasms disappear altogether. However, even after a short rest, muscle hypertonicity manifests itself with the same force.
In adulthood, a patient diagnosed with Thomsen's myotonia may look like an athlete if the disease affects the muscles of the limbs and trunk. Due to constant overexertion, muscle mass increases, the muscles hypertrophy and look large and pumped up.
The patient's muscles react with strong excitement even with weak external stimuli. Thus, a light blow to the affected muscles can lead to their hypertonicity. At the site of the touch, a tense muscle roll appears, which takes time to relax.
A myotonic attack is most often observed in two cases: in the initial phase of voluntary movement, requiring the participation of diseased muscles, and when they are exposed to cold. However, there are other provoking factors: prolonged stay in a static position, a sharp loud sound, an emotional outburst.
Diagnostics
To identify the disease, they use the methods of questioning, examination, palpating and tapping, as well as laboratory and instrumental diagnostics.
- Survey: identify the age of onset of the disease, its characteristic manifestations, dependence on hypothermia, family history;
- Examination: athletic build, detection of muscle hypertrophy;
- Palpation: muscle weakness in combination with dense ridges;
- Tapping: myotonic phenomenon - when tapping on a muscle roller, it contracts with prolonged relaxation;
- Tendon reflexes: not impaired;
- Biochemical blood test: increased level of creatine phosphokinase (an enzyme that destroys muscle protein);
- Genetic examination: identification of gene mutation and confirmation of the dominant nature of inheritance;
- Electromyography: multiple contractions of muscle cells up to 80 times per second (myotonic response). The frequency of contractions is constantly changing, which is accompanied by the hum of the apparatus (a symptom of a “dive bombardier”).
REFERENCE. As a rule, the clinical picture and electromyography data are sufficient to make a diagnosis. In difficult cases, a biopsy is performed, which reveals elongation and uneven expansion of muscle fibers. X-rays of bones also help in diagnosis (they determine osteoporosis and thinning of the periosteum).
Diagnosis of pathology
It is most often possible to diagnose myodystrophy based on the results of a survey of parents. If this disease is suspected, a physical examination is performed.
Taking blood tests to establish a diagnosis is mandatory
An important component of a comprehensive examination to obtain a complete picture is taking blood for analysis. Based on its results, the level of creatine phosphokinases is determined. This enzyme is also present in healthy muscles, but with muscular dystrophy its level increases significantly.
Physical examination includes electromyography. Based on its results, the electrical activity of the muscles can be determined. Structural abnormalities of muscle tissue are determined by taking a small sample of it for examination using a biopsy. According to its results, in patients with myodystrophy, not only a structural disorder is determined, but also an increased content of fat cells.
Echocardiography is required to detect signs of damage to the heart muscle. Diagnostics must be comprehensive in order to detect any lesion.
Treatment
Pathogenetic therapy is being carried out. There are no methods that can affect the genetic mutation, so etiological treatment has not been developed. Treatment is prescribed to each patient individually after consultation with a doctor.
Drug therapy
Medication therapy is not excluded in any case.
Most often prescribed drugs are from the following groups:
- Anticonvulsants – help relax contracted muscles and relieve stiffness. Diphenin, carbamazepine, and novocainamide are used strictly in moderate anticonvulsant doses;
- Diuretics that affect the permeability of cell membranes. Helps restore intracellular metabolism. From this group, diacarb is used according to the scheme;
- Ion channel blockers – prevent the development of tonic contractions. Mexiletine is used according to the scheme;
- Calcium antagonists – prevent the destruction of myofibrillar fibers and also have a cardioprotective effect. Nifedipine and disopyramide are used 3 times a day according to the scheme.
The course of treatment with each drug is 3 weeks, after which the doctor determines the break time and treatment is continued.
Physiotherapy
Almost always, the doctor prescribes physiotherapeutic exercises, but on their own they are not able to improve the situation:
- Galvanic currents to the area of the pathological focus;
- Electrophoresis with Actovegin and other vascular agents;
- Physiotherapy exercises under the supervision of a physician;
- Massage of the affected area (all types of massage are allowed, except tonic).
REFERENCE. The congenital nature of the pathology causes insufficient effectiveness of therapy. For severe complaints, glucocorticosteroids are used in short courses (5-7 days), which prevent the destructive effect of calcium ions.
Treatment of Thomsen's myotonia
You cannot count on a complete cure. The main goal of drug and physiological therapy will be to relieve symptoms and achieve stable remission. For this purpose, the patient is prescribed:
- Mexiletine is a sodium channel blocker that significantly reduces muscle hypertonicity;
- Diakarb - improves membrane permeability;
- Quinine - reduces muscle excitability, increases the refractory period;
- Diphenin is an effective anticonvulsant;
- Diuretics - maintain ionic balance, maintaining magnesium levels and reducing potassium.
Long-term use of medications has negative aspects: all medications have a wide range of side effects.
Non-drug therapy helps improve metabolic processes in muscle tissue and reduce its tonic spasms:
- exercise therapy;
- Electrophoresis;
- Acupuncture;
- Therapeutic swimming.
What is the prognosis for recovery?
The prognosis for life is relatively favorable. Complete recovery is impossible, but the disease progresses over a long period of time. In some patients, deterioration does not occur throughout life. Without treatment, ability to work is reduced.
With full-fledged therapy, improvement occurs in 90% of cases, and the ability to work is maintained for life. There is evidence of long-term remission under the influence of anticonvulsants. With age, muscle spasms weaken in all patients.
Useful video:
Possible complications
The long course of the disease leads to the involvement of many organs and systems in the process.
Their defeat occurs indirectly - due to prolonged muscle spasm:
- Spinal curvature: forward (lordosis), backward (kyphosis), sideways (scoliosis);
- Intervertebral hernia;
- Sleep apnea syndrome (suffocation during sleep);
- Cardiomyopathy (heart damage develops because the myocardium is represented by striated muscles);
- Muscular atrophy;
- Persistent changes in facial expressions, chewing and swallowing;
- Bruxism.
REFERENCE. Course therapy allows one to avoid complications and achieve long-term remission, but patients often develop addiction to the anticonvulsant. In this regard, the possibility of self-medication and long-term use of the same drug without medical supervision should be excluded.
Complications of pathology
Due to dystrophic processes in the muscles, even when the pathology is localized in a certain muscle group, the entire musculoskeletal system is gradually involved in the pathological process. Typically, patients are very susceptible to respiratory tract infections due to the involvement of the chest muscles in the pathological process. In the later stages of muscular dystrophy, respiratory infections can pose a fatal threat to a person.
Over time, thickening of the heart muscle develops. This affects the functioning of the entire cardiovascular system. The contractility of the heart muscle decreases.
Conclusion
Thomsen's myotonia is a rare neuromuscular disease. If you notice characteristic symptoms in a child, you should seek help and follow certain recommendations:
- Eliminate provoking factors (hypothermia);
- Do not self-medicate;
- Do not use medications outside the regimen or without medical advice;
- Provide the patient with peace and long sleep;
- Provide assistance when moving, going up and down stairs.
The congenital nature of the pathology makes it possible to identify it at an early age and begin complex treatment, which the patient may require throughout his life.