Tuberous sclerosis
(
sclerosis tuberosa
; lat. tuberosus tuberous; sclerosis; synonym:
Bourneville disease, Bourneville-Pringle disease
) is a hereditary progressive disease from the group of phakomatoses, characterized by combined damage to the nervous system, skin, eyes, bone and endocrine systems, and internal organs.
First described in 1863 by F. Recklinghausen. In 1880, D.-M. Bourneville, having identified this disease as a separate nosological form, introduced the term “tuberous sclerosis” and described in detail early neurol. symptoms, skin changes, etc. In 1890, JJ Pringle studied in detail adenomas of the sebaceous glands of the facial skin and emphasized that they are characteristic of Tuberous sclerosis.
According to Penrose (LS Penrose), T. s. in the general population it occurs with a frequency of approximately 1:600,000, among patients with varying degrees of mental retardation - 1:30,000.
Pathological anatomy
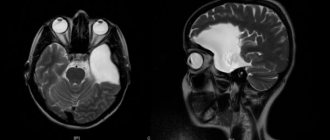
Fig.1.
Macroscopic specimen of the right hemisphere of the brain (medial surface) in tuberous sclerosis: arrows indicate multiple tubercles in the cerebral cortex and lateral ventricle. The brain in tuberous sclerosis is usually enlarged compared to the age norm, but microcephaly is sometimes observed (see). The cerebral cortex in individual areas of varying sizes is whitish-yellowish in color and has a greater density than the surrounding tissue. In the center of the areas of the altered cortex there are shallow depressions into which the vessels are immersed. On sections of the cerebral hemispheres, various numbers of mushroom-shaped tumor-like formations, or tubercles, are found in the cortex (Fig. 1); Their identification formed the basis for the name of the disease. Single or multiple grayish tubercles ranging in size from a pinhead to several centimeters in diameter are also found in the white matter of the cerebral hemispheres, in the lateral ventricles, more often near the thalamic ribbon, less often in the anterior and inferior horns of the lateral ventricles, in the aqueduct of Sylvius (aqueduct of the midbrain, T .) and in the fourth ventricle. In some cases, T. s. There are also changes in the cerebellum. Tumor-like formations can disrupt the outflow of cerebrospinal fluid from the ventricles and lead to the development of internal hydrocephalus (see). These formations are usually calcified, with foci of calcification located more often in the depths of the tubercle.
Rice. 2. Microscopic specimen of the cerebral cortex in tuberous sclerosis: the arrow indicates a large atypical neuron; impregnation according to Bielschowsky; x 400. Fig. 3. Microscopic specimen of the white matter of the cerebral hemisphere in tuberous sclerosis: 1 ~ large atypical cell; 2 — nested accumulation of cells with wrinkled cytoplasm and hyperchromic nuclei: hematoxylin-eosin staining; x 400.
Histological examination of “tuberous” areas in the cerebral cortex reveals atypical, bizarrely shaped large neurons (Fig. 2), the axons of which are often directed to the surface of the cortex, intertwined into balls, and embedded in the walls of blood vessels. Large atypical cells with one or two bubble-like nuclei are also found in the cortex, the cytoplasm of which, when stained with thionin, acquires a pale blue color. These cells, as established by modern immunological studies, are also neurons, and not astrocytes, as previously assumed. In the cerebral cortex there are also disturbances of normal cytoarchitecture with the absence of neurons, their atrophy and vacuolization. The cerebral cortex is not clearly demarcated from the white matter, it contains pronounced gliosis (see), argentophilic formations, granular balls located along the vessels (some researchers associate the appearance of granular balls with destructive processes caused by frequent epileptic seizures). The walls of the brain vessels are thickened, there are signs of fibrosis and hyalinosis (see). The soft and arachnoid membranes of the brain are thickened, and the number of melanophores in the latter is increased. In the tubercles located in the white matter of the cerebral hemispheres and cerebellum, single large atypical cells with a vesicular nucleus are found, as well as nested clusters of cells with hyperchromatic nuclei and wrinkled cytoplasm (Fig. 3). Tumor-like formations in the brain of patients I.e. In some cases they contain large round cells, similar to atypical neurons, in others - round or spindle-shaped cells with oval or elongated nuclei or multinucleated cells. These formations according to gistol. similar in structure to neuromas (see), subependymal astrocytomas (see), ependymomas (see). Nodules found in the eyeball are most often gliomas. Focal calcifications in the choroid of the eyeball, its angiomas (see), and malformations of the eye are also described.
In the myocardium of patients with T. s. tumor-like nodes are also found, often located in the region of the conduction system of the heart or growing in the cavity of the heart and having the structure of lipomas (see), fibrolipomas, rhabdomyomas (see). There are known observations of myocardial rhabdomyomatosis (accumulations of atypical cells in the myocardium). In the kidneys with T. s. detect small and giant hamartomas (see), capillary angiomas, as well as other malformations of blood vessels and various parts of the nephron. Also described are lipomyomas and “sarcomatous tissue” in the lymph nodes, lipomyomas, angiomyolipomas (see Kidneys) and angiomatous adenomas (see Angiomatosis) in the liver and spleen, cysts and myomatous formations in the lungs, accumulations of fatty and atypical epithelial cells in the thyroid gland, aplasia and hypoplasia of the testicles, lipomatous formations in the adrenal glands, adenomas (see) from the cells of the pancreatic islets, accumulations of stratified squamous epithelial cells in the anterior lobe of the pituitary gland, cysts in its posterior lobe, etc.
Causes of tuberous sclerosis
Bourneville disease is genetic in nature and is caused by mutations in the TSC1 and TSC2 genes, and therefore patients experience uncontrolled growth of tumor tissue.
There are 2 types of tuberous sclerosis depending on the location:
- Mutation of gene 34 locus of the ninth chromosome. In this case, there is a violation of the coding of hematin, an antioncogene that ensures the prevention of tumor transformation of cells;
- A mutation in the 13th region of the sixteenth chromosome leads to malfunctions of tuberin, a protein that blocks uncontrolled cell growth.
Expert opinion
Author: Andrey Igorevich Volkov
Neurologist, Candidate of Medical Sciences
Tuberous sclerosis is a very rare genetic disease characterized by the development of benign tumors in various organs. TS is diagnosed during puberty. The approximate frequency is 1 in 6000. If one of the parents has this disease, then the chance that it will be detected in the child is 50%. At the same time, there are statistics showing that 2/3 of cases are new mutations.
The clinical manifestations of the disease are very diverse and depend on the location of the tumors. Lesions of the central nervous system block the flow of nerve impulses, lead to delayed cognitive development, and provoke convulsions and spasms. Tuberomas can grow and block the flow of cerebrospinal fluid, causing unilateral hydrocephalus. Angiolipomas (kidney tumors) and polycystic kidney disease provoke the development of arterial hypertension. Skin lesions are very common.
Major and minor signs are used for diagnosis. To make a diagnosis of tuberous sclerosis, two major signs or one major sign in combination with two (or more) minor signs are sufficient. Blood and urine tests, kidney ultrasound, ECG, EEG, CT, MRI, and genetic studies are required.
Clinical picture
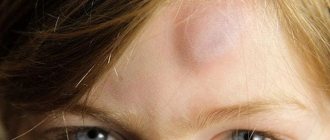
Rice.
4. Face of a patient with tuberous sclerosis: multiple adenomas of the sebaceous glands are visible. The disease develops in children 1-3 years old, usually begins with the occurrence of epileptic seizures (see Epilepsy) of a polymorphic nature: small, large, with generalized or focal seizures (see). Petty epileptic seizures of various forms (absences, myoclonic, akinetic, propulsive) as the disease develops turn into grand mal seizures with loss of consciousness. Focal motor, sensory seizures, and convulsive seizures of the type of Jacksonian epilepsy occur (see). Early symptoms are also mental retardation (see Mental retardation) and behavioral disturbances. The decline in intelligence reaches the level of imbecility and even idiocy. There is a clear correlation between mental decline and the severity of epileptic seizures. The neurological status reveals lesions of the cranial (cranial, T.) nerves, most often the oculomotor, trochlear and abducens nerves, pyramidal hemiparesis (see Hemiplegia), extrapyramidal hyperkinesis (see), etc. Along with this, hypothalamic disorders may occur: vegetative-trophic, endocrine-metabolic (diabetes mellitus, premature puberty, hypothyroidism, dysfunction of the pituitary-adrenal system). Skin manifestations occur later than neuropsychiatric disorders. For T. s. Characteristic is the appearance of adenomas of the sebaceous glands (see Adenoma of the sebaceous glands), located symmetrically in the shape of a butterfly on the face, mainly in the area of the wings of the nose, on the cheeks, and chin (Fig. 4); sometimes adenomas can spread to the neck and shoulder area. Eyes can be single or multiple, have the appearance of small or large papules of yellow or pink color, dense, reminiscent of sago grains. Along with adenomas, café-au-lait spots are found on the skin (see Melanosis), dense areas of “shagreen skin”, angiofibromas (see), angiectasia (see), hyper- or hypopigmentation (see Pigmentation). Angiofibromas and angiectasias can also occur on mucous membranes. In patients with T. s. dysembryogenetic changes in the skeletal system are often detected, for example, spina bifida (see). Tumor-like formations and cysts found in internal organs (heart, liver, kidneys) may clinically appear for a long time or not at all, or manifest themselves as nonspecific symptoms of damage to a given organ, for example, hematuria (see) with kidney damage, etc.
An ophthalmological examination reveals specific changes in the fundus: in the area of the optic nerve head - single nodular hamartomas or multiple gray-yellow nodules resembling mulberries; in the retina - changes in the pigment layer. Progression of pathol. changes in the eyes lead to glaucoma and blindness.
The course of the disease is progressive: central paralysis appears and increases (see Paralysis, paresis), extrapyramidal, hypothalamic, neuroendocrine, and mental disorders. The disease progresses especially rapidly during puberty.
Changes in the visual organs
benign tumors of the retina and optic nerve (often multiple) - retinal hamartomas of astrocytic origin. It is observed in 50% of patients. The tumor is difficult to notice at the very beginning.
It calcifies quite quickly. Ophthalmic examination should be undertaken as early as possible in children with a dilated optic papilla. The tumor is often located near the optic disc.
Calcifications in retinal hamartomas on both sides.
Diagnosis
The diagnosis is based on the appearance of epileptic seizures of a polymorphic nature in early childhood, mental retardation, delayed development of motor and speech functions, the presence of adenomas of the sebaceous glands on the face and other characteristic skin changes, changes in the fundus of the eye, progression of the disease, and the familial nature of the disease. When radiography of the skull reveals osteosclerosis (see) mainly of the cranial vault, signs of intracranial hypertension and petrification (see) in the structures of the brain, which in appearance resemble foci of calcification in toxoplasmosis and cytomegaly. Correct and early diagnosis is helped by computed tomography data (see Computer Tomography), which detects systemic changes in the brain, kidneys, liver, heart, etc. Computed tomography of the brain reveals multiple tumor-like formations, dilation of the ventricles, hydrocephalus of varying severity, petrification, which are localized in the cortex, most often in the frontal and parietal lobes, in the third and lateral ventricles. X-ray examination of the spine, pelvic bones, and limbs reveals foci of osteosclerosis, thickening of the periosteum, cyst-like formations in the periosteum of the bones of the arms and legs, osteoporosis (see) metacarpal and metatarsal bones, etc.
An electrophysiological study reveals focal or diffuse EEG changes of an epileptoid nature (see Electroencephalography). Cerebrospinal fluid is usually not changed, except in cases where hydrocephalus, cerebral edema, status epilepticus and other conditions complicating the course of the disease develop in severe brain lesions.
Differential diagnosis
carried out with neurofibromatosis (see), encephalotrigeminal angiomatosis (see), Hippel-Lindau disease (see Hippel-Lindau disease), Klippel-Trenaunay syndrome (see Blood vessels, malformations), Albright syndrome (see Pseudohypoparathyroidism), syndrome Larsen and malformations of c. n. With.
Diagnostics
In addition to collecting anamnesis, conducting an external and neurological examination, neuroimaging diagnostic techniques (MRI, MSCT) are additionally performed. Numerous foci of calcification are identified throughout the brain. Most of the formations are adjacent to the ventricular system.
Also, if a disease is suspected, an EEG study is performed. The EEG determines gross diffuse disturbances, sometimes there are also individual pronounced localized foci of epileptiform activity in the form of delta and theta waves, sharp waves, peaks, and peak-wave complexes.