Pineoblastoma is a fairly rare type of neoplasm that originates in or near the pineal gland. This tumor is a rare cerebral tumor. Pineoblastoma of the brain occurs most often in children; boys are usually affected by this pathology, although precise data on gender distribution is not available.
Table of contents
- What are embryonal, non-rhabdoid brain tumors and pineoblastoma?
- What are the types of tumors?
- How common are these types of tumors in children?
- Why do children get embryonal, non-rhabdoid brain tumors and pineoblastomas?
- What are the symptoms of the disease?
- How is the disease diagnosed?
- How is a treatment plan made?
- How are these types of tumors treated?
- What protocols and registries are used to treat children?
- What are the chances of being cured from these types of tumors?
What are embryonal, non-rhabdoid brain tumors and pineoblastoma?
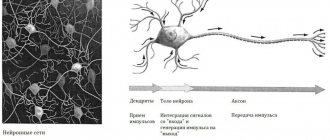
Embryonic, non-rhabdoid tumors of the central nervous system, which until recently were called “primary neuroectodermal tumors of the central nervous system (abbreviated as PNET-CNS; the term PNET-CNS was also used) and pineoblastomas are tumors that arise from mutated cells [cell] of the brain [brain] or spinal cord [spinal cord]. Because these types of tumors grow in the central nervous system [central nervous system], they are also called primary CNS tumors. That is, these are not metastases from other malignant tumors that grew in other organs and their cancer cells penetrated into the central nervous system.
Both embryonal, non-rhabdoid brain tumors and pineoblastoma arise from completely immature and undifferentiated [undifferentiated] cells of the central nervous system (specialists usually call them embryonic [embryonic]). And that's why they grow very quickly. These types of tumors are very similar in the structure of the tumor tissue when examined under a microscope (histological analysis). In this respect they also have similarities with medulloblastoma, an embryonal tumor of the cerebellum.
Embryonic, non-rhabdoid brain tumors and pineoblastomas differ from each other in that they grow in different parts of the central nervous system. Embryonic, non-rhabdoid tumors appear most often above the cerebellar tent [cerebellar tent] (specialists in this case use the term supratentorial), usually in the cerebral hemispheres [cerebrum]. That is why earlier experts used the term supratentorial PNET to distinguish them from such an embryonic tumor as medulloblastoma, which at that time belonged to the group of primitive neuroectodermal tumors, that is, PNET. But in rare cases, embryonal, non-rhabdoid tumors can grow in other parts of the central nervous system. Pineoblastomas, on the other hand, grow in the region of the pineal gland (pineal region; the pineal gland is a small part of the diencephalon [diencephalon]).
Both types of tumors grow aggressively. Often embryonic brain tumors grow from one cerebral hemisphere to the other, and/or move into the meninges, and continue to grow there. Pineoblastomas can also move from the pineal gland to other regions of the spinal cord and brain. Metastases [metastasis] beyond the central nervous system, i.e. in the bones, bone marrow, lungs or lymph nodes are very rare.
Therapy for pineoblastoma
If previously conservative treatment of patients was most often carried out, the use of microsurgical equipment today allows doctors to increase the radicality of tumor removal and increase the list of indications for surgical intervention. The modern treatment regimen is divided into 3 basic stages:
Surgery . Contraindications to surgical intervention are: involvement of the brain stem and choroid plexuses in the oncological process with the risk of massive bleeding during the operation. Bypass surgery is used as a palliative method and to stabilize the patient’s health before radical surgery.
Radiation therapy . Due to the high malignancy of pineoblastoma, general irradiation of the spinal cord and brain and additional irradiation of the tumor bed are performed. The use of capsules with radioisotopes for injection into the brain is the most effective method of radiation therapy with minimal risk of side effects.
Polychemotherapy . It is carried out in conjunction with radiation therapy after neurosurgery or as palliative treatment of inoperable tumors. Chemotherapy can be administered in several ways - when the drug is taken orally or injected into a muscle or vein, in this case it enters the bloodstream and affects tumor cells throughout the body (this method is called systemic chemotherapy). When the drug is injected directly into the organ, the spine, the drug affects the tumor cells in these areas (this is called local chemotherapy).
Combination chemotherapy involves the use of more than one antitumor agent. The choice of chemotherapy administration method and type of chemotherapy depends on the form and stage of the cancer. Most often, polychemotherapy is used, which consists of a combination of 2-3 cytostatic drugs (Etoposide, Vincristine, Carboplatin). The side effects of chemotherapy that occur require additional symptomatic treatment.
Would you like to receive an estimate for treatment?
*Only upon receipt of data on the patient’s disease, a representative of the clinic will be able to calculate an accurate estimate for treatment.
The choice of treatment methods depends on the following points:
- localization of the tumor in the brain;
- the patient's age at the time of diagnosis of the tumor;
- the number of tumors remaining after the operation;
- spread of cancer to other parts of the central nervous system: spinal cord and meninges, other organs - bones and lungs;
- whether it is a primary tumor or a relapse of the disease.
The most effective method of treatment is surgery, but if the tumor is very widespread, it is almost never performed.
The main goal of surgery is the absolute removal of pineoblastoma . Brain surgery is characterized by its complexity; the neurosurgeon must cut off the modified tissue so that healthy and functioning brain structures are not affected.
Some cancer treatments can cause side effects that may appear months or years after treatment in patients who have recovered. Such consequences are called long-term. These types of consequences of disease therapy include:
- various physical disorders.
- transformation of thinking, learning abilities, changes in memory, changes in mood, emotional state.
- The emergence of secondary cancer (new types of cancer).
Some long-term consequences can be cured or their symptoms reduced.
Video on the topic:
What are the types of tumors?
The group of embryonal, non-rhabdoid brain tumors (prior to the adoption of the new updated WHO classification [WHO classification] was called PNET, or PNET of the central nervous system) includes different types of tumors. They differ from each other in their histological characteristics (that is, in how the tumor tissue looks under a microscope) and in their molecular features.
According to the new World Health Organization (WHO) classification of tumors of the central nervous system, this includes primarily “embryonic tumors with multilayered rosettes” (experts use the English abbreviation ETMR for “embryonal tumor with multilayered rosettes”). Depending on whether there is a certain genetic change on chromosome 19 (the so-called C19MC amplification) or not, tumors are divided into “embryonic tumors with multilayered rosettes and damage to the C19MC gene” (accepted by experts the abbreviation in English is “ETMR C19MC-altered”) and for “embryonic tumors with multilayer rosettes without diagnostic specifications” (specialists use the English abbreviation “ETMR NOS”). For other tumors from this group, for example, the extremely rare medulloepithelioma and other embryonal, non-rhabdoid tumors of the central nervous system, there are no other specifications (specialists in this case say “tumors are not specified”).
Until recently, many researchers classified pineoblastomas and embryonal, non-rhabdoid tumors into the same group, since they are rare and there are similarities in how the diseases progress and how they are treated. However, accumulated experience has shown that pineoblastomas are quite different at the molecular level from other embryonal tumors of the central nervous system and therefore they must be considered as a separate type of tumor.
Embryonic, non-rhabdoid brain tumors and pineoblastomas are classified as high-grade tumors, so-called Grade IV tumors in the WHO classification.
How common are these types of tumors in children?
Embryonic, non-rhabdoid tumors of the central nervous system and pineoblastoma are extremely rare in children and adolescents. Of all types of brain tumors in children and adolescents, they make up about 2%. In Germany, approximately 10 children and adolescents under 15 years of age (new cases) become ill with these types of tumors per year. Embryonic tumors of the central nervous system mainly appear in the first years of life. Middle age is children between three and four years old. The ratio of sick boys and girls is approximately the same. Pineoblastoma mainly occurs in children and young adults.
Leading clinics in Israel
Assuta
Israel, Tel Aviv
Ikhilov
Israel, Tel Aviv
Hadassah
Israel, Jerusalem
In its structure, neoplasia is similar to other malignant neoplasms that originate in tissues of neuroectodermal origin: medulloblastoma, retinoblastoma. The cancer process originates in the cells of the pineal gland, a gland that produces melatonin (it regulates the cycles of wakefulness and sleep). The pineal gland synthesizes several bioactive substances - hormones that affect human biorhythms and participate in the activity of the endocrine, nervous and digestive systems.
This disease occurs in only 0.1% of cases of the total number of intracranial tumors; according to ICD10, pineoblastoma has code C75.3.
The pineal gland, or pineal gland, has minimal dimensions and is located between the hemispheres of the brain behind the 3rd ventricle. Next to it is the “Sylvian aqueduct” through which cerebrospinal fluid circulates. This complex anatomical zone where pineoblastoma develops is called the pineal region.
Macroscopically, pineoblastoma is a densely elastic formation of a brown hue; microscopically, this tumor is a collection of small monomorphic cells with hyperchromatic nuclei.
Several features of pineoblastoma can be distinguished:
- Aggressive growth. This leads to the appearance of a pronounced clinical picture in just a few months.
- Increased production of melatonin, a hormone that is responsible for normal falling asleep and waking up.
- Blockage of the ventricles of the brain during tumor growth, which leads to hydrocephalus.
- Spread of the tumor to the spinal cord.
- The predominant diagnosis of this neoplasia is in children and adolescents up to approximately 18 years of age.
Why do children get embryonal, non-rhabdoid brain tumors and pineoblastomas?
Embryonic tumor of the central nervous system and pineoblastoma occur when cells of the nervous tissue [nervous tissue] begin to change malignantly (that is, mutate). So far, no one knows exactly why this process begins in the first place. It is known that the risk of developing a brain tumor increases if the child has previously had to undergo brain radiation. This stage must be passed through, for example, in the treatment of acute leukemia or eye cancer (retinoblastoma).
In addition, new data has emerged that show that in cancer cells themselves there are certain changes in genes or chromosomes [chromosomes]. As a result of such changes, cell development and cellular communication are disrupted. And this may be the reason why a healthy cell turns into a cancerous one. Since these types of cancer are very rare, specialists have only sporadic data on possible typical molecular genetic [molecular genetic] changes.
Good to know: in extremely rare cases, the appearance of pineoblastoma is associated with hereditary retinoblastoma (the so-called trilateral retinoblastoma, a form of eye cancer). In this case, the tumor appears due to genetic [genetic] changes in the so-called retinoblastoma gene. For more information about trilateral retinoblastoma, see our text on retinoblastoma in children.
Clinic
At the time of clinical presentation, almost all patients have HCF with typical symptoms (H/P, vomiting, drowsiness, memory impairment, abnormal increase in head circumference in infants, seizures). There may be Parinaud's syndrome (or Sylvian aqueduct syndrome). Boys with choriocarcinoma or germinoma with scintiotrophoblastic cells may experience precocious puberty as a result of the action of βhCG secreted in the cerebrospinal fluid, similar to the action of LH. Metastasis to the cerebrospinal fluid may cause radiculopathy and/or myelopathy
What are the symptoms of the disease?
Typically, in children and adolescents with embryonal tumor or pineoblastoma, symptoms of the disease appear very quickly, as the tumor itself grows quickly and uncontrollably. Just like other types of brain cancer, the symptoms of the disease depend primarily on the age of the child, as well as on which specific part of the central nervous system the brain tumor grew in and how far it has already spread throughout the body. It is customary for specialists to divide symptoms into general (doctors talk about nonspecific symptoms) and local (doctors talk about specific symptoms).
Nonspecific symptoms (general)
General symptoms appear regardless of where exactly the tumor has grown. They generally appear in other diseases that are not related to tumors of the central nervous system. These may be, for example, headaches and/or back pain, dizziness, loss of appetite, nausea and vomiting (a typical symptom in brain cancer, when a person vomits at all without depending on food intake [vomiting on an empty stomach]; very often this occurs due to lying down in the morning), weight loss, increased fatigue/fatigue, decreased academic performance, loss of concentration, changes in a person's character and behavior, developmental delays.
Most often, these symptoms appear because pressure begins to slowly build up inside the skull. Compression of the internal structures in the skull can be caused by the growing tumor itself, and/or due to the tumor, the free circulation and outflow of cerebrospinal fluid (CSF) is disrupted. Due to a violation of the outflow of cerebrospinal fluid, the child may develop water on the brain (hydrocephalus). If dropsy of the brain appears in infants and young children, when the fontanelles have not yet closed, then they can see how much the head has grown in volume (macrocephaly).
Specific symptoms (local)
Specific (local) symptoms indicate exactly where in the central nervous system the tumor has grown and the functioning of which control centers it disrupts. For example, if a tumor grows in the cerebrum [cerebrum], or in the diencephalon [diencephalon], the child may have motor problems (paralysis) and/or seizures. Also, the child may experience disturbances in vision, sleep, and normal behavior; mood swings and problems regulating appetite may occur. A patient with pineoblastoma may experience gaze paresis. Experts call this form of visual impairment Parinaud's syndrome. Due to the fact that the pineal gland tumor grows in a certain place in the diencephalon, it compresses the center there, and among other things, the eyeballs cannot move upward.
Good to know: if a child has one of these symptoms, or several at once, this does not mean that he has an embryonal tumor, pineoblastoma, or another form of brain cancer. Many of the symptoms mentioned can appear in other diseases, relatively less insidious, which have nothing to do with brain tumors. However, we recommend that if you have certain complaints, you should consult a doctor as soon as possible (for example, if your child has constant headaches, or in young children the volume of the head quickly becomes disproportionately large) in order to find out the exact cause of these symptoms. If doctors really find some type of brain cancer in a child, then they need to begin treating the disease as quickly as possible.
Causes of pineoblastoma tumors and its symptoms
The causes that lead to low-quality tumors of the cerebral system are constantly being studied. Experts in the treatment of brain cancer abroad claim that one of the provoking factors for the formation of pineoblastoma is the mutation of the RB1 gene. There are also other reasons that can lead to a mutagenic process:
- different types of ionizing radiation, increased radiation;
- exposure to carcinogenic substances;
- pathologies during pregnancy;
- head injury;
- complications after viral diseases;
- heredity.
The symptoms of pineoblastoma are very diverse, especially in pediatric patients. Its manifestation is influenced by the location, size of neoplasia, and the age of the child.
At the initial stage of the disease, symptoms are expressed as follows:
- regular headaches;
- nausea and vomiting;
- vision problems;
- elevated temperature;
- weight fluctuations;
- epileptic seizures.
In young children, pineoblastoma can cause anxiety and moodiness. With further growth of neoplasia and its penetration into the midbrain, strabismus and double vision develop. If the pathological process affects the cerebellum, then body coordination is impaired. Problems with melatonin production lead to changes in the biorhythms of a sick child.
Due to compression of the brain as a result of significant enlargement of the ventricles in a limited space, intracranial pressure increases.
In addition, with the development of pineoblastoma in adolescence, early puberty is observed due to impaired synthesis of reproductive hormones.
In addition to the listed indicators of pineoblastoma of the brain, other deviations in normal health are possible. This is apathy, weakness, lethargy.
When it comes to cancer treatment abroad, doctors try to take into account that with pineoblastoma the symptoms may be identical to other types of tumor-like formations of the central nervous system, so they carry out differential diagnosis.
How is the disease diagnosed?
If the child’s medical history (history) and the results of an external examination [external examination] give the pediatrician a suspicion of a malignant tumor in the central nervous system, then the doctor refers him to a clinic that specializes in pediatric and adolescent oncology (clinic of pediatric oncology and hematology). Because if such a tumor is suspected, then a full examination is carried out by specialists of various profiles. First, they must confirm the diagnosis of whether the child actually has a malignant tumor of the central nervous system [brain tumor]. Secondly, if the diagnosis is confirmed, they must say what specific type of tumor the child has and how far the disease has spread throughout the body. Only by answering these questions can we optimally plan treatment tactics and give a prognosis for the disease.
What tests and studies are done to confirm the diagnosis?
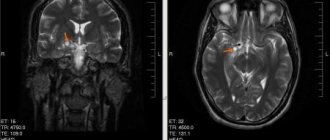
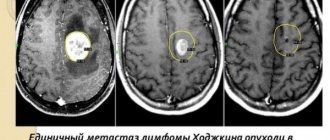
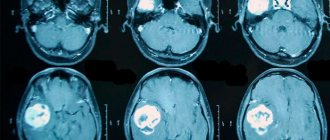
To check whether the child has an embryonic tumor of the central nervous system or pineoblastoma, the medical history is carefully studied again and an external examination is performed. To make an accurate diagnosis, doctors first need to do diagnostic imaging - magnetic resonance imaging (MRI), sometimes also CT, that is, computed tomography. Using these methods, you can accurately determine whether there is a tumor in the brain and whether it has managed to metastasize into the spinal canal. In the pictures you can see exactly where the tumor has grown, what size it is, where the borders of the tumor with neighboring structures are. They can also be used to accurately see whether the child has hydrocele.
To definitively confirm the diagnosis, a piece of tumor tissue (biopsy) is needed. She is sent for histological and molecular genetic analysis. As a rule, during an operation to remove a tumor, a part of the tumor is taken and it is this that is sent for this diagnosis. The volume of histological studies and, first of all, molecular genetic analyzes has grown greatly in recent years. Modern laboratory research methods are able to describe the molecular genetic properties of tissue so accurately that this makes the diagnosis more accurate. Depending on the results of this diagnosis, specialists also receive information about how the disease will progress (disease dynamics), for example, how the tumor is able to grow and how quickly. Already today, based on the results of molecular genetic studies, doctors decide on what plan they will use to treat the child. In the future, the value of this information for modern diagnostics will become even greater.
Diagnostics to determine the extent of the disease
If the diagnosis of embryonal tumor of the central nervous system or pineoblastoma is confirmed, then specialists conduct additional tests and studies to understand how much cancer cells have spread throughout the central nervous system. For example, MRI images of the entire central nervous system (that is, images of the brain and spinal cord) show (that is, visualize) those metastases that doctors call macroscopic metastases (those that are visible on the images). Additionally, specialists must conduct a microscopic examination of the cerebrospinal fluid (CSF) in the spinal canal. Because it may contain cancer cells that are not visible on MRI images. Most often, cerebrospinal fluid is taken after surgery, when a puncture is made in the lower back/lumbar spine (lumbar puncture). There is a cavity from which it is easiest to obtain cerebrospinal fluid.
What tests and studies are done before starting treatment?
Before treatment, children may additionally undergo other tests and studies. For example, they do an ECG (electrocardiogram) and an echocardiogram (echocardiogram) to check how the heart is working. Various blood tests are also done to check the general condition of the child’s body, as well as to assess how well individual organs (for example, kidneys and liver) are working, or whether there may be any metabolic disorders. These baseline data are then regularly compared with the results obtained during treatment. If any changes occur during treatment, it is easier for specialists to correctly assess the situation.
Diagnosis of pineoblastoma
To make a diagnosis, anamnesis is first collected in order to have an idea of the course of pregnancy, the process of childbirth, the postpartum period, early symptoms and the time of onset of the disease. Further examination of the patient consists of:
- biochemical blood test (an increase in ALT, AST, a decrease in protein concentration is determined);
- general blood test (signs of leukocytosis and anemia may be noticeable);
- blood tests for tumor markers;
- examination by an ophthalmologist. If swelling of the optic nerve head is detected, conclusions can be drawn about increased intracranial pressure.
- cerebrospinal fluid studies. This helps eliminate the effect of infection on the central nervous system. Significant cytosis, increased protein concentration, and the presence of cancer cells are also diagnosed. Red blood cells can be detected in the cerebrospinal fluid during hemorrhage.
- neuroimaging. Most often this is an MRI of the brain; in older people (if there are contraindications to MRI), a cerebral CT can be performed. This helps to identify neoplasia, establish the growth pattern, the extent of the process and its location.
- stereotactic tumor biopsy. It is used when deciding whether neurosurgical treatment is necessary.
- histological examination of the biopsy specimen.
In addition to the above examination methods, before surgery, neurosurgeons often conduct the following studies: cerebral angiography or MR angiography, ventriculography.
It is imperative to carry out differential diagnosis with benign pineocytoma, encephalitis, other intracranial space-occupying formations (abscess, cerebral cyst, tumors), and traumatic injuries. In the first year of life, congenital brain pathologies are excluded. In young children, some symptoms (fever, nausea or vomiting) may be mistaken for symptoms of an intestinal infection.
How is a treatment plan made?
After the final diagnosis, a treatment plan is drawn up. In order to create the most individual treatment program, specifically selected for a particular patient, and to assess the possible risks of relapse of the disease (risk-adapted therapy), the team of treating doctors must take into account certain factors that influence the prognosis of the disease in a particular child (the so-called prognostic factors or risk factors).
Important prognostic factors are the information that was obtained after completing the entire diagnosis: what specific type/type of tumor was found in the child, where exactly the tumor is located, how much it has managed to grow (tumor size) and metastasize. In addition, the biological (molecular biological) characteristics of the tumor tissue (specialists can talk about the molecular profile of the tumor) increasingly determine which treatment is considered the most optimal in a particular case. The age of the child and his general health also matter. The age of the child at the time of diagnosis determines whether the child can be sent for radiation therapy or not. All these factors are included in the treatment plan in order to obtain the most effective treatment result for each patient.
How are these types of tumors treated?
Children with embryonal brain tumors or pineoblastoma should be treated only by doctors from children's clinics specializing in pediatric oncology. It is there that highly qualified specialists (doctors, nurses) with a specialization in pediatric oncology work and are proficient in modern therapy programs. In these hospitals, doctors of different specialties belong to different working groups, which are constantly in close contact. Together they create treatment plans, discuss and manage their patients. Therapy programs are regularly improved. Their goal is to treat the child as gently as possible, that is, with minimal side complications and long-term consequences.
Treatment of children with embryonal brain tumor or pineoblastoma consists of surgery, courses of chemotherapy and, depending on the age of the child, radiation therapy (irradiation).
Operation
The first step in the treatment of children with an embryonal tumor of the central nervous system or pineoblastoma is surgery. Its goal is complete removal of the tumor without “microscopically visible” remains. This means that after surgery, tumor remnants are not visible under a neurosurgical microscope. At the same time, the neurosurgeon strives, as far as possible, not to damage/affect healthy brain tissue. However, it is often impossible to completely remove an embryonal tumor of the central nervous system or pineoblastoma of the brain due to the complex location of the tumor (that is, the specific place where it grew).
When the tumor is removed, in most children the outflow of cerebrospinal fluid (CSF) is restored if it was disrupted. If the child had dropsy of the brain (hydrocephalus), then even before surgery to remove the tumor, additional surgery may be required to normalize the outflow of cerebrospinal fluid. Some children have a permanent drainage system installed.
Non-surgical treatment stage
Since embryonic tumors of the central nervous system and pineoblastomas of the brain grow into neighboring tissues (experts call in this case “infiltrative tumor growth”), and they often diverge along the cerebrospinal fluid pathways (cerebrospinal fluid system) to other parts of the central nervous system, then one treatment the visible part of the tumor is not enough. Therefore, after the operation, the stage of non-surgical treatment begins. This stage consists of radiation therapy [radiation] and/or courses of chemotherapy [chemotherapy]. During chemotherapy courses, children receive drugs that block cell growth (cytostatics). The goal of this treatment is to stop the growth of cancer cells or destroy them. Radiation therapy is done using high-energy electromagnetic [electromagnetic] radiation. It is applied externally through the skin to the region that needs to be irradiated. The radiation destroys the DNA of cancer cells and they begin to die.
The decision on how exactly the child will be treated (what treatment methods the doctors will choose, how intense the courses of chemotherapy/radiation therapy will be) depends on the age of the patient, what specific type of tumor was found in the child, what molecular biological features the tumor has, and also depends on whether the child has metastases. In addition, they take into account whether it was possible to completely remove the tumor during the operation.
How are children with pineoblastoma of the brain treated?
Usually, after the maximum possible removal of the tumor, children over 4 years of age, in whom pineoblastoma has not had time to metastasize, receive radiation therapy to the entire central nervous system (in this case, doctors use the term craniospinal irradiation). And then they additionally irradiate the tumor region. After the radiation stage, children receive courses of so-called maintenance chemotherapy. They use several different cytostatics. If pineoblastoma has managed to metastasize, treatment becomes more intense. For example, radiation therapy is performed with a higher dose of radiation, and before it begins, the child additionally receives a course of so-called induction chemotherapy.
In children under 4 years of age, the development of brain tissue has not yet been fully completed. Therefore, doctors refuse radiation therapy or seek to postpone it to a later date in order to minimize the risks of serious long-term complications. Instead of this stage of treatment, children after surgery receive courses of chemotherapy consisting of several drugs. Some children may be given courses of high-dose chemotherapy [high-dose chemotherapy] to increase their chances of being cured of the disease. And after it, specialists perform an autologous bone marrow transplantation [autologous bone marrow transplantation].
How are children with other types of embryonal brain tumors treated?
New information has accumulated on other types of embryonal tumors of the central nervous system. Researchers were able to identify many new subgroups. Therefore, experts strive to individualize treatment, which is planned depending on the characteristics of each of these subgroups.
It is important to know: how exactly a particular child will be treated is decided by the attending physician after a detailed discussion with the sick person or his relatives.
Die im Anschluss vorgestellten Therapieoptionen basieren auf Empfehlungen der Studien-/Registerzentrale (gemäß HIT-MED-Guidance) und erheben an dieser Stelle keinen Anspruch auf Vollständigkeit. Insbesondere für Patienten unter vier Jahren gibt es keine Standardtherapie-Empfehlung. Wie die Behandlung beim einzelnen Patienten genau abläuft, entscheidet der behandelnde Arzt im Gespräch mit den Patienten beziehungsweise deren Angehörigen.
Treatment
The optimal treatment strategy for pineal region tumors still needs to be determined.
Hydrocephalus
The best treatment for patients admitted for acute HCF is external ventricular drainage (EVD). It allows fluid control, prevents peritoneal contamination, and, in a significant number of patients, avoids the need for a permanent shunt system, which is no longer required after surgical removal of the tumor (although ≈90% of patients with germ cell tumors require a shunt). It is important to have access to the ventricle in the postoperative period in case of acute HCF (through the NVD or Fraser burr hole).
Stereotactic techniques
Can be used to establish a diagnosis (biopsy). Caution is required during the procedure, because this area is rich in vessels (vein of Galen, basal vein of Rosenthal, internal cerebral veins, posterior medial choroidal artery), which may be displaced from their normal location.
Complication rate during stereotactic interventions: mortality ≈1.3%, complications ≈7%, 1 case of contamination per 370 patients. Diagnosis rate ≈94%.
A limitation of stereotactic biopsy is that it may not reveal the histological heterogeneity of some tumors. The study showed that there is an association between intervention trajectory and the occurrence of complications. Therefore, they recommend the inferior frontal approach, which passes below the internal cerebral veins. However, another study did not confirm this connection. But they found that complications occurred more often with solid tumors (pineocytoma, teratoma, astrocytoma). Therefore, if difficulties are encountered when first attempting to penetrate the tumor to take a biopsy, they recommend using an open approach. SRS can be used to treat some of these lesions.
Outcomes of surgical interventions Mortality: ≈5-10%. Postoperative complications: new visual field impairment, epidural fluid accumulation, infection, cerebellar ataxia
What protocols and registries are used to treat children?
In Germany, almost all children and adolescents with embryonal tumors of the central nervous system or pineoblastoma of the brain, as well as with recurrent disease, are treated according to standardized protocols called therapy optimization studies and treatment registries. German protocols, or therapy optimization studies, are clinical studies and are strictly controlled. Their goal is to treat sick children using the most modern developments. At the same time, these studies provide an opportunity to improve treatment approaches and thereby achieve progress in treatment.
Children who are not being treated according to the current research protocol (for example, if at the time of illness the old protocol was closed and a new one has not yet opened; or if the sick person does not meet the criteria that are mandatory for admission to the current protocol), go through treatment registries. Treatment registries are created and operate in order to advise all patients from modern scientific positions. Also, to ensure high quality of treatment, the research team of a particular protocol usually develops detailed therapeutic recommendations. And when attending physicians contact them, they advise them on choosing the optimal therapy for each individual child.
In Germany, in 2011, the long-term HIT 2000 protocol ended its work, according to which children and adolescents with embryonal, non-rhabdoid tumors of the central nervous system (they were previously called central nervous system-PNET), or patients with pineoblastomas (as well as patients with medulloblastomas and ependymomas) were treated. Numerous clinics throughout Germany and Austria have been using this protocol. Currently, a new protocol is not open for children and adolescents who are newly diagnosed with embryonal, non-rhabdoid brain tumor or pineoblastoma. But these patients are registered in the I-HIT-MED therapeutic registry.
Currently in Germany they operate according to the following registers:
- I-HIT-MED Therapeutic Registry : Children with embryonal tumors of the central nervous system (formerly PNET-CNS) or pineoblastoma, who are currently or in the future not eligible for inclusion in a research protocol, can be treated according to the recommendations of this therapeutic registry (the name of the registry is an abbreviation from English International HIT-MED Registry , that is, the international HIT-MED registry). Patients receive an individual treatment program, that is, it depends on the specific form of the disease. In order for a child to be included in the I-HIT-MED therapeutic register, it does not matter what type of treatment will be carried out. Evaluating the effectiveness of a particular type of treatment is not the purpose of the registry. The therapeutic register is managed by the HIT-MED research protocol team at the University Hospital Hamburg (Hamburg-Eppendorf) (research team leader Prof. Stefan Rutkowski, MD).
- Therapeutic registry HIT-REZ : this registry opened in January 2015. The opportunity to enter it exists for those children who become ill for the first time, but the disease does not respond to treatment; or if the disease has returned to a child who has already been treated (relapse). The treatment regimens in this registry do not test any new therapies or medications. However, the registry is the responsibility of the research office. And he makes therapeutic recommendations that are based on the current results of German anti-relapse protocols (for example, the results of the HIT-REZ 2005 research protocol, which closed in 2021) and international anti-relapse protocols. The central research office is located at the Center for Child and Adolescent Medicine at the University Hospital Essen (headed by Prof. Dr. Gudrun Fleischhak).
What are the chances of being cured from these types of tumors?
According to the German Children's Cancer Registry (Deutsches Kinderkrebsregister), the chances of recovery (prognosis) in children and adolescents with an embryonal, non-rhabdoid tumor (the old name PNET-CNS) are about 60% (in medical statistics, 5-year survival rates are accepted). The prognosis for overall survival of children with pineoblastoma is slightly better.
However, in each specific case, the prognosis of the disease depends on many factors. Firstly, the group of diseases that includes embryonal tumors of the central nervous system is, as experts say, heterogeneous. This means that the chances of recovery are different for everyone and it depends on the specific type of tumor. In addition, the stage of the disease and the age of the child play a special role in assessing the prognosis. For example, children and adolescents with a metastatic form of the disease, as a rule, have a more unfavorable prognosis than those with a localized form (that is, a tumor without metastases). Also, in young children who cannot yet receive radiation therapy for treatment, the prognosis is unfavorable. Their chances of long-term survival are estimated by experts to be from 20 to 30%.
A necessary note: when we call the percentage of recovered children with an embryonal tumor of the central nervous system or with pineoblastoma, this means that we are giving only accurate statistics for this form of brain cancer in children. But no statistics can predict whether a particular child will recover or not.
When we say that a child has been cured, this must be understood as “no tumor.” Because modern treatment methods, although they give such a result when the tumor is no longer there, the tumor itself can leave behind certain damage in the body, and the treatment itself can lead to certain consequences and complications for a long time. Therefore, after treatment, children should receive medical care for a long time and, if necessary, intensive rehabilitation [rehabilitation].
Prognosis and prevention for pineoblastoma
With such a complex disease as pineoblastoma, the prognosis can be ambiguous due to serious consequences. Despite the fact that the cost of cancer treatment abroad is quite expensive, combination therapy in foreign clinics helps prolong the patient’s life.
Important factors in predicting the disease are the degree of progression of the pathology and the age criterion. Children over 6 years of age have a more favorable prognosis than younger patients, since pineoblastoma cells in patients in infancy have an increased degree of malignancy.
In addition, the predisposition of a tumor-like neoplasm to spread metastases leads to a significant percentage of relapses, but in operated patients the five-year survival rate is about 60%.
Since the exact causes of pinaeocyte mutations have not been established, preventing pathology is quite problematic. However, at the slightest suspicion of brain cancer, it is necessary to consult with specialized doctors and undergo an examination. The patient can choose oncology centers abroad based on different criteria for geolocation, price, rating and seek medical assistance in the treatment of pineoblastoma.
Bibliography:
- Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Anual Report 2021 (1980-2017). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2021 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 22605/ 0/ 2df4719687ba2596d4216218a4f4632763b64847/ jb 2018s.pdf] KAA2019
- Fleischhack G, Rutkowski S, Pfister SM, Pietsch T, Tippelt S, Warmuth-Metz M, Bison B, van Velthoven-Wurster V, Messing-Jünger M, Kortmann RD, Timmermann B, Slavc I, Witt O, Gnekow A, Hernáiz Driever P, Kramm C, Benesch M, Frühwald MC, Hasselblatt M, Müller HL, Sörensen N, Kordes U, Calaminus G: ZNS-Tumoren. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie. Springer-Verlag GmbH Deutschland, 2. vollständig überarbeitete Auflage 2021, 359 [ISBN: 978-3-662-43685-1] FLE2018
- Mynarek M, Pizer B, Dufour C, van Vuurden D, Garami M, Massimino M, Fangusaro J, Davidson T, Gil-da-Costa MJ, Sterba J, Benesch M, Gerber N, Juhnke BO, Kwiecien R, Pietsch T, Kool M, Clifford S, Ellison DW, Giangaspero F, Wesseling P, Gilles F, Gottardo N, Finlay JL, Rutkowski S, von Hoff K: Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: analysis of pooled European Society for Pediatric Oncology (SIOP-E) and US Head Start data. Neuro-oncology 2017 Apr 1; 19:576 [PMID: 28011926] MYN2017
- Rutkowski S, Trollmann R, Korinthenberg R, Warmuth-Metz M, Weckesser M, Krauss J, Pietsch T: Leitsymptome und Diagnostik der ZNS-Tumoren im Kindes- und Jugendalter. Gemeinsame Leitlinie der Gesellschaft für Neuropädiatrie und der Gesellschaft für Pädiatrische Onkologie und Hämatologie 2021 [URI: https://www.awmf.org/uploads/tx_szleitlinien/ 025-022l_S1_ZNS-Tumoren_Kinder_Jugendliche_20 16-09.pdf] RUT2016
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW: The 2021 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica 2021, 131: 803 [PMID: 27157931] LOU2016
- Gerber NU, von Hoff K, Resch A, Ottensmeier H, Kwiecien R, Faldum A, Matuschek C, Hornung D, Bremer M, Benesch M, Pietsch T, Warmuth-Metz M, Kuehl J, Rutkowski S, Kortmann RD: Treatment of children with central nervous system primitive neuroectodermal tumors/pinealoblastomas in the prospective multicentric trial HIT 2000 using hyperfractionated radiation therapy followed by maintenance chemotherapy. International journal of radiation oncology, biology, physics 2014, 89: 863 [PMID: 24969797] GER2014
- Bode U, Zimmermann M, Moser O, Rutkowski S, Warmuth-Metz M, Pietsch T, Kortmann RD, Faldum A, Fleischhack G: Treatment of recurrent primitive neuroectodermal tumors (PNET) in children and adolescents with high-dose chemotherapy (HDC) and stem cell support: results of the HITREZ 97 multicentre trial. Journal of neuro-oncology 2014, 120: 635 [PMID: 25179451] BOD2014
- Friedrich C, von Bueren AO, von Hoff K, Gerber NU, Ottensmeier H, Deinlein F, Benesch M, Kwiecien R, Pietsch T, Warmuth-Metz M, Faldum A, Kuehl J, Kortmann RD, Rutkowski S: Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro-oncology 2013, 15: 224 [PMID: 23223339] FRI2013
- Timmermann B, Kortmann RD, Kühl J, Rutkowski S, Meisner C, Pietsch T, Deinlein F, Urban C, Warmuth-Metz M, Bamberg M: Role of radiotherapy in supratentorial primitive neuroectodermal tumor in young children: results of the German HIT- SKK87 and HIT-SKK92 trials. Journal of clinical oncology 2006, 24: 1554 [PMID: 16575007] TIM2006
Embryonic tumors of the central nervous system and pineoblastoma (brief information) (443KB)